

Образование и утилизация кетоновых тел
источников. Двумя основными видами ацетоновых тел являются ацетоацетат и -гидроксибутират.-гидроксибутират - это восстановленная форма ацетоацетата. Ацетоацетат образуется в клетках печени из ацетил~КоА. Образование происходит в

митохондриальном матриксе.
Первоначальная стадия этого процесса катализируется ферментом - -кетотиолазой. Затем ацетоацетил-КоА конденсируется со следующей молекулой ацетил-КоА под влиянием фермента ГОМГ-КоА синтетазы. В результате образуется-гидрокси--метилглютарил-КоА. Затем фермент - ГОМГ-КоА лиаза катализирует расщепление ГОМГ-КоА на ацетоацетат и ацетил-КоА. В дальнейшем ацетоуксусная кислота восстанавливается под влиянием фермента-гидроксибутиратдегидрогеназы и в результате образуется-оксимасляная кислота. Количество ацетоацетата, которое восстанавливается в-гидроксибутират, зависит от соотношения НАДН/НАД+. Восстановление это происходит под влиянием фермента-гидроксибутиратдегидрогеназы. Печень служит главным местом образования кетоновых тел благодаря высокому содержанию ГОМГ-КоА синтетазы в митохондриях гепатоцитов.
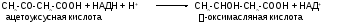
Ацетон образуется из ацетоуксусной кислоты при декарбоксилировании. Из печени поток кетоновых тел попадает во внепеченочные ткани.

Обратите внимание: эти реакции происходят в митохондриях. В цитозоле имеются изоферменты --кетотиолазы и ГОМГ~КоА синтетазы, которые также катализируют образование ГОМГ~КоА, но в качестве промежуточного продукта в синтезе холестерола. Цитозольный и митохондриальный фонды ГОМГ~КоА не смешиваются.
Образование кетоновых тел в печени контролируется состоянием питания. Такое контрольное действие усиливается инсулином и глюкагоном. Принятие пищи и инсулин снижают образование кетоновых тел, в то время как при голодании стимулируется кетогенез вследствие увеличения количества жирных кислот в клетках. При голодании усиливается липолиз, растет уровень глюкагона и концентрация цАМФ в печени. Происходит фосфорилирование, тем самым активация ГОМГ-КоА синтетазы. Аллостерическим ингибитором ГОМГ-КоА синтетазы выступает сукцинил-КоА.
В норме кетоновые тела являются источником энергии для мышц; при продолжительном голодании они могут использоваться центральной нервной системой. Следует иметь ввиду, что окисление кетоновых тел не можетпроходить в печени. В клетках других органов и тканей оно протекает в митохондриях. Такая избирательность обусловлена локализацией ферментов, катализирующих этот процесс.
Сначала -гидроксибутират дегидрогеназа катализирует окисление-гидроксибутирата до ацетоацетата в НАД+-зависимой реакции. Затем с помощью фермента, сукцинилКоА:АцетоацетилКоА трансферазы, кофермент А перемещается с сукцинилКоА на ацетоацетат. Образуется ацетоацетилКоА, который является промежуточным продуктом последнего витка-окисления жирных кислот. Этот фермент в печени не образуется. Именно поэтому там не может происходить окисление кетоновых тел. Зато спустя несколько суток после начала голодания в клетках мозга начинается экспрессия гена, кодирующего этот фермент. Тем самым мозг адаптируется к использованию кетоновых тел в качестве альтернативного источника энергии, снижая свою потребность в глюкозе и белке Тиолаза довершает расщепление ацетоацетил-КоА встраивая КоА по месту разрыва связи междуиуглеродными атомами. В результате образуется две молекулы ацетил-КоА.
Интенсивность окисления кетоновых тел во внепеченочных тканях пропорциональна их концентрации в крови. Общая концентрация кетоновых тел в крови обычно ниже 3 мг/100 мл, а средняя ежесуточная экскреция с мочой составляет приблизительно от 1 до 20 мг. В определенных метаболических условиях, когда происходит интенсивное окисление жирных кислот, в печени образуются значительные количества так называемых кетоновых тел.
Состояние организма, при котором концентрация кетоновых тел в крови выше нормальной, называется кетонемией. Повышенное содержание кетоновых тел в моче называется кетонурией. В тех случаях, когда имеет место выраженная кетонемия и кетонурия, в выдыхаемом воздухе ощущается запах ацетона. Он обусловлен спонтанным декарбоксилированием ацетоацетата в ацетон. Эти три симптома - кетонемия, кетонурия и запах ацетона при дыхании объединяются общим названием - кетоз.
Кетоз возникает в результате недостатка доступных углеводов. Например, при голодании их мало поступает (или не поступает) с пищей, а при сахарном диабете, вследствие недостатка гормона - инсулина, она не может эффективно окисляться в клетках органов и тканей. Это приводит к дисбалансу между этерификацией и липолизом в жировой ткани в сторону интенсификации последнего. В результате большое количество жирных кислот поступает в кровоток, а затем - в клетки. Эти кислоты являются главным субстратом для образования кетоновых тел в печени. Поскольку в результате их -окисления образуется ацетил-КоА, естественно, что при увеличении количества окисляемых жирных кислот возрастает доля синтезируемых кетоновых тел.
Гуморальная регуляция синтеза и распада триацилглицеролов в жировой ткани тесно связана с проблемой ожирения. Это состояние характерно, прежде всего, для жителей развитых стран, у которых на 100% выше вероятность преждевременной (раньше, чем средняя продолжительность жизни) смерти; у людей с избыточным весом эта вероятность выше на 10% - 25%.
Формула развития ожирения проста. Оно развивается тогда, когда поступление в организм энергосубстратов превышает расходование энергии. Среди причин развития этого состояния, включающих генетические и средовые факторы, важнейшее значение имеют состав принимаемой пищи и специфические регуляторы энергетического баланса. Одним из таких соединений является продукт экспрессии так называемого гена ожирения (ob - гена) - лептин (leptos – греч. «тонкий»).
Лептин является белком, состоящим из 167 аминокислот. Местом синтеза лептина являются адипоциты, в гораздо меньшей степени - эпителиальные клетки желудка и плаценты. Причем, чем больше в этих клетках накапливается триацилглицеролов, тем больше там образуется лептина. Образовавшись, белок секретируется адипоцитами и не депонируется в клетках. До настоящего времени неизвестны механизмы, регулирующие экспрессию ob генов. По всей вероятности, к этому процессу имеют отношение глюкокортикоиды и инсулин.
|
|
Нормальная мышь (справа) и мышь с ожирением (слева), у которой имеется ob/ob мутация. Много лет назад генетики выявили у мышей два вида рецессивных мутаций. Наличие их в гомозиготном состоянии приводит к значительному ожирению. Гены получили название obиdb. Вес мышей с мутациями этих генов в три раза превышает вес нормального животного, а масса жира - в 5 раз. У них также появляются признаки диабета, непереносимости холода, подавления иммунитета, отставания в половом созревании. |

Действие лептина на депонирование триацилглицеролов в жировой ткани опосредовано его влиянием на гипоталамические центры, контролирующие поведение и чувство голода, температуру тела и энергозатраты. Такое его влияние опосредовано взаимодействием со специфическими рецепторами. Рецептор для лептина кодирует ген – db. Клонирование гена осуществили в 1995г. Он имеет в своем составе 1 трансмембранный домен. Экспрессия рецептора осуществляется в мозге (гипоталамус, хориоидное сплетение), Т-лимфоцитах, эндотелиальных клетках сосудистой стенки, меньше – в легких и почках. Он обнаружен у мышей и у человека.
|
|
|
|
. Схематическое изображение механизма действия лептина, направленного на снижение веса | |


Другими участниками процесса регуляции массы тела являются:
Нейропептид Y. Синтезируется во многих областях мозга; самый мощный из известных на сегодняшний день стимуляторов аппетита; снижает расход энергии; лептин подавляет аппетит, ингибируя экспрессию нейропептида Y.
Меланокортины. Воздействуют на некоторые гипоталамические нейроны и ингибируют потребление пищи.
Карбоксипептидаза Е (жировой ген) – фермент-протеаза процессинг проинсулина и др. гормонов (нейропептида Y)
Митохондриальные разобщающие белки – обнаружены в буром жире, белом жире и мышечных клетках. Они разобщают процессы тканевого дыхания и окислительного фосфорилирования, тем самым энергия, которая выделяется в ходе тканевого дыхания расходуется на теплопродукцию, а не на анаболические процессы в виде АТФ.
Бета-адренергические рецепторы – присутствуют в жировой ткани; связывание норадреналина с этим рецептором на поверхности адипоцитов усиление транскрипции митохондриальных разобщающих белков увеличенное теплообразование в результате окисления жирных кислот.
У человека, как оказалось, мутации ob и db генов редко являются причиной патологического ожирения. Вместе с тем, концентрация лептина в крови у таких людей обычно повышена, что позволяет думать скорее о какой-то потере чувствительности к лептину, но не о дефиците этого белка.
Министерство здравоохранения республики Беларусь
Учреждение образования
«Гомельский государственный медицинский университет»
Кафедра биохимии
Обсуждено на заседании кафедры _(МК или ЦУНМС)___
Протокол № 200 года_____
ЛЕКЦИЯ
По биологической химии
для студентов 2 курса лечебного факультета
Тема: Патология липидного обмена
Время 90 минут
Учебные цели:
1. Сформировать представление о причинах, вызывающих патологию липидного обмена
2.Сформулировать понятия- а)Холестериноз;б)Холестеринопатии; в)Липидозы
Литература:.
1.Биохимия человека., Р.Марри, Д.Греннер, П.Мейес, В.Родуэлл.- М.книга ,2004.- т.1-2.
2.Основы биохимии,А.Уайт, Ф.Хендлер,Э.Смит, Р.Хилл, И.Леман.-М. книга,1981,т. 1-.2,.с.
3.Наглядная биохимия,Кольман., Рем К.-Г-М.книга 2004г.
4.Липиды, липопротеиды и атеросклероз. А.Н.Климов, Н.Г.Никульчева, Питер., С-Петербург. 1995г.,-С. 298.
5. Холестериноз., Ю.М. Лопухин., А.И. Арчаков., Ю.А. Владимиров., Э.М.Коган., М.,Медицина.,-С.352.
Материальное обеспечение
1.Мультимедийная презентация
РАСЧЕТ УЧЕБНОГО ВРЕМЕНИ
|
№п/п |
Перечень учебных вопросов |
Количество выделяемого Времени в минутах |
|
1. |
Причины патологии липидного обмена |
|
|
2. |
Понятие холестериноза и холестеринопатии |
|
|
3. |
Атеросклероз и причины его вызывающие |
|
Всего 90 минут
Введение:
Патология липидного обмена возникает по двум причинам:
а) при нарушении переваривания и всасывания липидов;
б) при нарушении метаболизма липидов и липопротеидов.
В основе нарушения переваривания и всасывания липидов лежат три группы патологических процессов:1) в поджелудочной железе, сопровождающиеся дефицитом панкреатической липазы; 2) в печени- при закупорке желчных протоков и фистуле желчного пузыря, приводящие к дефициту желчи; 3) в кишечнике, сопровождающиеся снижением метаболической активности слизистой оболочки, где локализованы ферменты синтеза ТГ. В соответствии с этим различают панкреатическую, гепатогенную и энтерогенные формы стеаторей. Стеаторея- нарушение переваривания жиров.
Панкреатическая стеаторея вызывается дефицитом панкреатической липазы, что наблюдается при хроническом панкреатите, врожденной гипоплазии пакреас и муковисцедозе, когда наряду с другими железами поражена и поджелудочная железа.
Гепатогенная стеаторея наблюдается при врожденной атрезии желчных путей, механических желтухах, гепатитах, циррозе.
Энтерогенная стеаторея отмечается при целиакии, абеталипопротеинемии, интестинальной лимфангиэктазии, интестинальной липодистрофии, амилоидозе и обширной резекции тонкого кишечника.
При переваривании пищевых жиров в пищеварительном тракте высвобождаются моноглицериды и высшие жирные карбоновые кислоты (ВЖК)., которые после всасывания образуют ТГ. Последние являются основными компонентом хиломикронов (ХМ). Липиды, являясь гидрофобными , транспортируются кровью в виде особых надмолекулярных образований- липопротеидов(ЛП), в состав которых входят ХС, ТГ, ФЛ (фосфолипиды) и апобелки.
Диагностическое значение имеет определение в крови содержания ТГ, СЖК, ХС, желчных кислот, ФЛ и ЛП а также состав последних.
При переваривании пищи, в пищеварительном тракте высвобождается большое количество ТГ и ВЖК, которые после всасывания в слизистой кишечника образуют ресинтезированные липиды, Являясь экзогенными по своей природе, эти ресинтезированные ТГ, встраиваются в ХМ и через грудной лимфатический проток попадают в большой круг кровообращения. Это относится к ЖК, содержащим > 10-12 атомов С.Жирные кислоты с короткими цепями непосредственно попадают в портальный кровоток и далее в печень, где они наряду с эндогенными ( синтезированными в печени ТГ) встраиваются в пре-бетаЛП-ЛПОНП.
Холестерин- составная часть клеточных мембран и ЛП, особенно ЛПНП.
Независимо от происхождения, свободный ХС встраивается в липидные мицеллы, включающие СЖК, желчные кислоты и лизолецитин. . Мицеллы отдают свободный ХС клеткам слизистой оболочки кишечника, где экзогенный ХС смешивается с эндогенным и подвергается частичной эстерификации холестеринэстеразой. Далее ХС секретируется в лимфу, где появляется в составе ЛПОНП и ХМ. В крови он переходит из ЛПОНП в ЛПНП. Окисление ХС это его единственный путь его необратимого удаления из организма, т.е. из мембран и ЛП-комплексов., и этот процесс характерен не для всех типов клеток. Оксигеназные системы находятся в клетках печени и органов, синтезирующих стероиды.
Окисление ХС идет двумя путями: за счет биосинтеза желчных кислот и биосинтеза стероидных гормонов. Первый путь окисляет от 60-80% всего ежедневно, образующегося в организме ХС, второй путь- всего2-4 %. Монооксигеназный путь окисления при участии цитохрома Р450. Желчегенез ускоряется при гиперхолестеринемии и снижается у больных атеросклерозом.
Холестерин в патологии. В основу классификации холестериноза (накопления ХС) и холестеринопатий положен принцип содержания ХС в целом организме, крови, в отдельных органах
|
Содержание ХС в мембранах клеток |
Содержание ХС в крови |
Содержание ХС в отдельных органах, тканях |
|
Повышенное содержание ХС в мембранах-неосложненное течение-физиологическое старение |
Дислипопротеинемия-изменение соотношения классов липопротеидов |
Липидозы ( Болезнь Нимана-Пика, болезнь Тея- Сакса и т.д.) |
|
Осложненное течение -атеросклероз |
Гиперлипопротеинемия (ГЛП), классификация ВОЗ |
|
|
Пониженное содержаниеХС в мембранах: а)канцерогенез;б)вирусные инфекции |
Гипопротеинемия |
|
Тип 1. ( изменение содержания ХС в организме)
1.Холестериноз:- накопление ХС
а) неосложненный ( физическое старение, старость, смерть)- накопление ХС в плазматических мембранах, как результат снижения стероидогенеза ( секс-гормонов);
б) осложненный (атеросклероз ) в форме ИБС ( инфаркт миокарда, кардиосклероз), ишемии мозга ( инсульт, тромбоз), ишемии конечностей ( гангрена), ишемии органов и тканей, дегенерации брадитрофных структур ( катаракта, остеохондроз), связанный с уменьшением желчегенеза.
2. Дефицит ХС:
А) злокачественные новообразования, сопровождаются гипоХС, с низким содержанием ХС в плазматических мембранах клеток.;
Б) вирусные инфекции сопровождаются гипоХС, повышенной проницаемостью плазматических мембран для вирусов
ТИП 2. Дислипопротеинемии-изменение соотношения классов ЛП в крови. – это гипо и гиперхолестеринемии. Это первичные наследственные гипо и приобретенные вторично состояния. ( Классификация по Фредриксону)
ТИП 3. Накопление ХС в отдельных органах и тканях
Атеросклероз. В этиологии атеросклероза ведущая роль принадлежит риск-факторам, в частности артериальной гипертензии, эмоциональной лабильности, курению, гиперТГ-демии. Сферу этих факторов можно ограничить, если выделить три категории больных, в одной из которых влияние этих факторов в развитии атеросклероза действительно велико, а в другой несущественно.
Первая группа- это лица, устойчивые к развитию атеросклеротических изменений.. Содержание ХС у них в крови меньше 5.2 ммоль/л и частота атеросклротических изменений МИНИМАЛЬНА. К этой группе относятся долгожители с наследственной гипобеталипопротеидемией ( отсутствие ЛПНП в крови) или семейным дефицитом ЛПВП. Даже полное отсутствие ЛПВП не приводит к развитиюатеросклеротических изменений сосудов эласто-мышечного типа. Действие факторов риска на эту популяцию не приводит к развитию атеросклероза.
Вторая группа-это люди с содержанием ХС от 5.2-до 9 ммоль/л, т.е. основная часть взрослого населения. Влияние факторов риска в этой группе является решающимпри образовании атеросклеротических изменений и появления атеросклероза любой локализации. Большую роль в диагностике заболеваний играет холестериновый индекс атерогенности, когда высокая концентрация ХС в ЛПНП, на фоне низкого содержания ЛПВП является плохим прогностическим признаком. Отдельные факторы риска влияют на развитие атеросклероза в различной степени.
Третью группу составляют лица с концентрацией ХС в плазме выше, чем 9 ммоль/л. Это больные атеросклерозом и отсутствие атеросклеротических изменений сосудов при такой концентрации ХС в крови встречается крайне редко. Следует отметить, что подавляющее большинство людей принадлежит ко 2 группе. Поэтому ограничение сферы деятельности других факторов за исключением ХС, определнной популяции людей, переводит стероид из категории способствующих, в категорию причинных.

Появление и развитие атеросклеротических изменений рассматривается с позиций инфильтрационной теории, согласно которой основным действующим фактором, вызывающим образование и развитие атеросклеротических изменений в стенке сосуда является ХС ЛПНП.Этот класс ЛП подвергается значительным изменениям в зависимости от питания, условий внешней и внутренней среды. Меняется не только липидный, но и апопротеиновый состав, заряд, жесткость ЛП- частицы. При этом заметно нарастает содержание иммуноглобулина LgG.
Апо-В содержащие ЛП (ЛПНП и ЛПОНП) образуют растворимые комплексы с ГАГ соединительнотканного матрикса аретрий, приводя к структурным изменениям стенки сосуда.


Строение артериальной стенки Дисфункция эндотелия
Такое изменение структуры и конформации ЛП вызывает активацию перекисного окисления липидов, входящих в состав ЛП. Экзогенные вещества (например жирорастворимые витамины), также как и перекиси липидддов, могут с одной стороны вызывать конформационные изменения апоаротеинов, а сдругой служить гаптенамит. Не исключено появление в составе ЛП фенотипов апопротеинов или их мутантных форм. Антитела к ЛП появляются в 2х случаях: в отвте на модифицированные in vivo ЛП, которые приобретают аутогенные свойства, и , во-вторых в ответ на воздейтсвие патогенных или других факторов, когда клетки иммунокомпетентной системы синтезируют антитела, образующие иммунные комплексы с нативными плазменными ЛП. В обоих случаях образуется иммунный комплекс ЛП-антитело, который рассматривается как модифицированный ЛП , иначе реагирующий с клеткой, чем нативный ЛП.
Физиологический смысл формирования иммунного комплекса необходим для более быстрого удаления антигена из крови. Иммунный комплекс ЛПНП-LgG захватывается макрофагами на 23% активнее, чем нативные ЛП. Избыточный захват иммунных комплексов ЛП- антитело макрофагами приводит к превращению последних в пенистые клетки, играющие важную роль в развитии атеросклеротичесмких поражений артерий ( ксантоматоз, регрессирование и стадии атеросклероза) Появление пенистых клеток во внутренней оболочке артерий является характерным морфологическим признаком прогрессирующего атеросклеротического процесса. Ускоренному образовании. Пенистых клеток способствует специфический набор рецепторов, у макрофагов интимы артериальной стенки. Это:
1.апо-В рецепторы к ЛПОНП и ЛПНП
2. рецепторы к ЛПОНП и ЛППП
3.рецепторы к комплексу ЛПОНП с ГАГ
4.рецепторы к комплексу белок- эф.ХС
5. рецепторы к иммунным комплексам ЛП-иммуногобулин LgG
6. рецепторы, связывающие модифицированные ЛП- сукцинилированные и ацетоацетилированные.
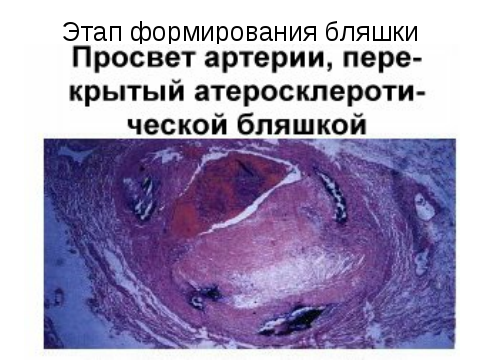
Гибель пенистых клеток приводит к выходу в межклеточное пространство липидов. Таким образом липидоз интимы происходит одновременно как за счет ЛП, поступающих их плазмы, так и за счет освобождения из пенистых клеток ХС. Накопление последних сопровождатеся очаговым склерозом, образованием пятен и полос, являющихся основой для фиброзных бляшек.
Осложненные поражения сопровождаются развитием воспалительных, дегенеративных и некротических изменений в атеросклеротических бляшках. Дальнейшее разрастание СТ, увеличение размеров бляшки, утолшение интимы, образование изъязвлений с выходом детрита в кровоток, геморрагии в бляшку, возникновение тромбов, кальциноз.

Существенную роль в появлении ишемического синдрома при атеросклерозе имеет поражение клеток крови. Увеличение содержания ХС в мембране эритроцитов приводит к изменению ее физико-химических свойств и нарушению функции мембранно-связанных ферментов. Одновременно с индексом ХС/ФЛ увеличивается микровязкость и скрость агрегации тромбоцитов.. При этом снижается активность Na+ K+ АТФ-азы, увеличиваются размеры эритроцитов, деформируемость клеток падает. Тромбоциты также накапливают ХС, скорость их агрегации также возрастает, мембрана становится более регидной. Такие же изменения характерны и для лимфоцитов.
Нарушение реологических свойств крови наряду с изменением микрососудов при холестеринозе обуславливает нарушение микроциркуляции и связанную с этим ишемию. Результатом этого является тромбоз, ишемия, склероз и некроз.
Генетические варианты первичных гиперлипопротеинемий
Табл.1.
|
Тривиальное название |
Тип по Fredricson |
Причина ГЛП |
Связь с атеросклерозом |
Распростаненность в популяции |
|
Семейная ГХС: гомозиготная гетерозиготная
|
II A |
Дефицит ЛПНП-рецепторов |
++++ ++ |
1: 1006 1: 500 |
|
Семейная ГХС, причина- дефект апо В-100 |
II A |
Нарушение катаболизма ЛПНП: дефект апоВ |
+++ |
1:500-1:6000 |
|
Семейная дисбета-ЛП
Семейная комбинированная ГЛП
|
III
IIA или IIB или IV |
Гомозиготный тип Е2/Е2, усиленное образование ЛПОНП , нарушение катаболизма ремнант
Повышенный синтез апо-В-100 и ЛПОНП
|
++
++ |
1: 5000
1: 100-1: 300
|
|
Полигенная ГХС |
IIA |
? |
+ |
? |
|
Семейная ГТГ |
IV |
Усиленное образование и замедленный клиренс ТГ и ЛПОНП |
+ |
1: 500 |
|
Семенная гиперХМ |
I или V |
Дефицит ЛПЛ или апо С-II |
-- |
1:106 |
|
Семейная гипер-ЛП(а)-емия |
|
Усиленное образование ЛП(а) |
+++ |
? |
|
Семейная гиперальфаЛП |
Повышенный уровень ЛПВП |
Усиленное образование апо А-1: дефицит-ЭХС: замедленный катаболизм ЛПВП |
- |
? |
Вторичные гиперлипидемии
Табл.2.
|
Заболевание или состояние |
Изменение уровня липидов |
ХМ |
ЛПОНП |
ЛПНП |
ЛПВП |
|
Диабет Гипотиреоз
Хроническая почечная недостаточность
Нефротический синдром
Холестаз
Алкоголь
|
Ув.ТГ Ув. ХС
Ув.ТГ
Ув.ХС Ув.ТГ
Ув.ХС
Ув.ТГ |
Ув-ы
Пов.
|
Ув-ы
Увеличены
Повышены
|
Ув.ХС
N или выше
Повышены
Повышен.
|
Понижены N или снижен
Сниженны
Снижены
Снижены
N или выше
|
доц.каф. Свергун В.Т.
Дата