

2.4.22. Посторонние жирные кислоты в маслах методом газовой хроматографии
Испытание на посторонние жирные кислоты проводят путем перевода жирных кислот, содержащихся в испытуемом масле, в метиловые эфиры.
МЕТОД А
Этот метод неприменим для масел, содержащих глицериды жирных кислот с эпокси-, гидроксиэпокси-, циклопропиловыми или циклопропениловыми группами,а также для масел, в составе которых большая часть жирных кислот имеет длину цепи менее восьми атомов углерода, и для масел с кислотным числом более 2,0.
Испытание проводят методом газовой хроматографии (2.2.28).
Испытуемый раствор. Испытуемое масло высушивают перед метилированием, если это указано в частной статье. 1,0 г масла помещают в круглодонную колбу вместимостью 25 мл со шлифом, снабженную обратным холодильником и газоотводной трубкой. В колбу прибавляют 10 мл метанола безводного Р, 0,2 мл раствора 60 г/л калия гидроксида Р в метаноле Р, присоединяют обратный холодильник и, пропуская через смесь азот Р со скоростью около 50 мл/мин, встряхивают и нагревают до кипения. Когда раствор станет прозрачным (обычно через 10 мин), продолжают нагревание в течение последующих 5 мин. Затем колбу охлаждают под проточной водой и содержимое переносят в делительную воронку. Колбу промывают 5 мл гептана Р, переносят смывы в ту же делительную воронку и встряхивают. Прибавляют 10 мл раствора 200 г/л натрия хлорида Р и энергично встряхивают. Оставляют до расслоения, затем переносят органический слой в сосуд, содержащий натрия сульфат безводный Р и через некоторое время фильтруют.
# Допускается применение других методик перевода содержащихся в испытуемом масле жирных кислот в метиловые эфиры, указанных в частной статье.
Раствор сравнения (а). Готовят 0,50 г смеси веществ, применяемых для калибровки (калибровочной смеси), состава, приведенного в одной из Табл. 2.4.22, как указано в частной статье (если в частной статье не указан определенный раствор, готовят смесь, состав которой приведен в Табл. 2.4.22.-1). Смесь растворяют в гептане Р и доводят объем раствора этим же растворителем до 50,0 мл.
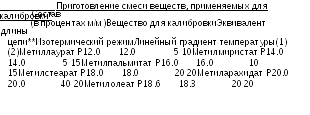
Табл. 2.4.22.-1
Табл.
2.4.22.-2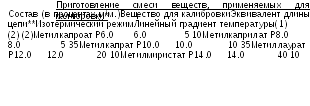
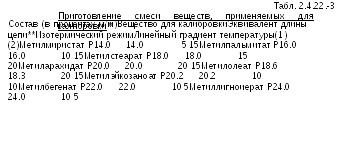 *Для
газовой хроматографии с применением
капиллярной колонки и делением потока
рекомендуется прибавлять к смеси
веществ, применяемых для калибровки,
компоненты с большей длиной цепи.
*Для
газовой хроматографии с применением
капиллярной колонки и делением потока
рекомендуется прибавлять к смеси
веществ, применяемых для калибровки,
компоненты с большей длиной цепи.
**Эти значения, вычисленные с использованием калибровочной кривой, даны в качестве примера для колонки, заполненной полиэтиленгликоль сукцинатом Р (1) и макроголом 20 000 Р (2).
Раствор сравнения (b). 1.0 мл раствора сравнения (а) доводят гептаном Р до 10.0 мл.
Хроматографируют на газовом хроматографе с пламенно-ионизационным детектором в следующих условиях:
колонка стеклянная или из нержавеющей стали длиной от 2 м до 3 м и внутренним диаметром от 2 мм до 4 мм, заполненная диатомитом носителем для газовой хроматографии Р с размером частиц от 125 мкм до 200 мкм, на который нанесено от 5% до 15% полиэтиленгликольсукцината Р или полиэтиленгликольадипината Р;
газ-носитель - азот для хроматографии Р;
скорость газа-носителя - 25 мл/мин;
температура колонки - 180°С;
температура инжектора и детектора - 200оС.
При необходимости или если указано в частной статье, температуру колонки увеличивают от 120°С до 200°С со скоростью 5°С в мин.
Хроматографировать возможно также на газовом хроматографе с пламенно-ионизационным детектором в следующих условиях:
колонка капиллярная стеклянная или кварцевая длиной от 10 м до 30 м и внутренним диаметром от 0.2 мм до 0.8 мм, внутренняя поверхность которой покрыта слоем поли[(цианопропил)(метил)][(фенил) (метил)]силоксана Р или макрогола 20 000 Р толщиной от 0,1 мкм до 0,5 мкм или другой подходящей неподвижной фазой;
газ-носитель - гелий для хроматографии Р или водород для хроматографии Р;
скорость газа-носителя - 1,3 мл/мин (для колонки с внутренним диаметром 0,32 мм);
деление потока - 1:100 или менее в зависимости от внутреннего диаметра применяемой колонки (в случае использования колонки с внутренним диаметром 0,32 мм деление потока должно составлять 1:50);
температура колонки - от 160°С до 200°С, в зависимости от длины колонки и используемой неподвижной фазы (для колонки длиной 30 м, покрытой слоем макрогола 20 000 Р, температура должна составлять 200°С);
температура инжектора и детектора - 250°С.
При необходимости или если указано в частной статье, температуру колонки увеличивают от 170°С до 230°С со скоростью 3°С в мин (для колонки, покрытой слоем макрогола 20 000 Р)
Хроматографируют 0,5 мкл раствора сравнения (а). Чувствительность системы регулируют таким образом, чтобы высота основного пика на полученной хроматограмме составляла от 50% до 70% шкалы регистрирующего устройства.
Определяют времена удерживания жирных кислот, входящих в состав калибровочной смеси. Хроматографируют 1 мкл раствора сравнения (b) и рассчитывают отношение сигнал/шум для пика, соответствующего метилмиристату.
Хроматографируют от 0,5 мкл до 1,0 мкл испытуемого раствора. Время хроматографирования должно в 2,5 раза превышать время удерживания метилолеата. Хроматограмму оценивают, как описанно ниже.
При использовании калибровочных смесей №1 или №3 хроматографическая система считается пригодной, если выполняются следующие условия:
на хроматограмме раствора сравнения (а) число теоретических тарелок (n) (2.2.28), вычисленное для пика, соответствующего метилстеарату, составляет не менее 2000 для набивной колонки и не менее 30 000 для капиллярной колонки;
на хроматограмме раствора сравнения (а) коэффициент разделения (Rs) (2.2.28) пиков, соответствующих метилолеату и метилстеарату, составляет не менее 1,25 для набивной колонки и не менее 1,8 для капиллярной колонки;
на хроматограмме раствора сравнения (b) отношение сигнал/шум (2.2.28) для пика метилмиристата составляет не менее 5.
При использовании калибровочной смеси №2 хроматографическая система считается пригодной, если выполняются следующие условия:
на хроматограмме раствора сравнения (а) число теоретических тарелок (n) (2.2.28), вычисленное для пика, соответствующего метилкапрату, составляет не менее 1500 для набивной колонки и не менее 15 000 для капиллярной колонки;
на хроматограмме раствора сравнения (а) коэффициент разделения (Rs) (2.2.28) пиков, соответствующих метилкаприлату и метилкапрату, составляет не менее 2 для набивной колонки и не менее 4 для капиллярной колонки;
- на хроматограмме раствора сравнения (b) отношение сигнал/шум (2.2.28) для пика метилкапроата составляет не менее 5.
ОЦЕНКА ХРОМАТОГРАММ
Следует избегать условий хроматографирования, которые могут дать неразделенные пики (наличие компонентов с небольшим различием между временами удерживания, например, линоленовая и арахидоновая кислоты.
Качественный анализ. Строят калибровочную кривую, используя хроматограммы раствора сравнения (а) и данные Таблиц 2.4.22.
а) Для хроматограмм, полученных в изотермическом режиме, вычисляют логарифмы приведенных времен удерживания как функцию эквивалента числа атомов углерода в жирных кислотах. Калибровочная кривая насыщенных кислот представляет собой прямую линию. Логарифмы приведённых времен удерживания ненасыщенных кислот расположены на этой линии как точки, соответствующие не целым значениям «эквивалента длины цепи». Идентификацию компонентов жирных кислот испытуемого масла проводят, рассчитываая логарифмы приведённых времён удерживания пиков, полученных на хроматограмме испытуемого раствора, и устанавливая по калибровочной кривой «эквивалентны длины цепи».
# Допускается идентификация жирных кислот испытуемого масла путем сравнения времен удерживания пиков на хроматограмме испытуемого раствора с временами удерживания пиков на хроматограмме раствора сравнения или на стандартной хроматограмме, описанной в частной статье.
# Приведенное время удерживания - разница между временем удерживания пика вещества и временем удерживания несорбирующегося (в условиях определения) вещества.
б) Для хроматограмм, полученных с использованием линейного градиента температуры, определяют времена удерживания, находящиеся в зависимости от числа атомов углерода в жирных кислотах, и идентифицируют жирные кислоты, входящие в состав испытуемого масла, путем сравнения с калибровочной кривой.
Количественный анализ. Обычно используют метод внутренней нормализации; при этом сумму площадей всех пиков на хроматограмме, кроме пиков, относящихся к растворителю, принимают за 100%. Рекомендуется применение электронного интегратора. Содержание каждого компонента вычисляют как отношение площади соответствующего пика к сумме площадей всех пиков. Пики, площадь которых составляет менее 0.05% от суммы площадей всех пиков, не учитывают, если нет других указаний в частной статье.
В определенных случаях, т.е. при наличии жирных кислот с 12 или менее атомами углерода, в частной статье должен быть указан поправочный коэффициент для преобразования площадей пиков в проценты (м/м).
МЕТОД В
Этот метод неприменим для масел, содержащих глицериды жирных кислот с эпокси-, гидроксиэпокси-, циклопропиловыми и циклопропениловыми группами и для масел с кислотным числом более
2,0.
Испытуемый раствор. 0,100 г испытуемого вещества помещают в центрифужную пробирку с завинчивающейся крышкой, растворяют в 1 мл гептана Р и 1 мл диметилкарбоната Р и энергично перемешивают при умеренном нагревании (от 50°С до 60°С). К еще теплому раствору прибавляют 1 мл раствора 12 г/л натрия Р в метаноле безводном Р, приготовленного с необходимыми предосторожностями, и энергично перемешивают в течение 5 мин. Прибавляют, 3 мл воды дистиллированной Р и энергично перемешивают в течение 30 с. Центрифугируют в течение 15 мин с ускорением 1500 g. Хроматографируют 1 мкл органического слоя.
Растворы сравнения и оценка хроматограмм. Если в частной статье нет других указаний, поступают, как указано в Методе А.
Хроматографируют на газовом хроматографе с пламенно-ионизационным детектором в следующих условиях:
кварцевая колонка длиной 30 м, с внутренним диаметром 0,25 мм, покрытая слоем макрогола 20 000 Р толщиной 0,25 мкм;
газ-носитель - гелий для хроматографии Р;
скорость газа-носителя - 0,9 мл/мин;
деление потока - 1:100;
температура колонки и скорость подъема температуры - по следующей программе:
|
|
Время (мин) |
1емпература (°С) |
Скорость подъема температуры (о С/мин) |
Примечания |
|
Колонка |
0-15 |
100 |
- |
Изотермический режим |
|
15-36 |
100^225 |
10 |
Линейный градиент температуры | |
|
36-61 |
225 |
- |
Изотермический режим | |
|
Инжектор |
|
250 |
|
|
|
Детектор |
|
250 |
|
|
МЕТОД С
Этот метод неприменим для масел, содержащих глицериды жирных кислот с эпокси-, гидроперекисными, альдегидными, кетоновыми, циклопропиловыми и циклопропениловыми группами и сопряженными полиненасыщенными и ацетиленовыми компонентами из-за частичного или полного разрушения этих групп.
Испытуемый раствор. 0,10 г испытуемого вещества помещают в коническую колбу вместимостью 25 мл, растворяют в 2 мл раствора 20 г/л натрия гидроксида Р в метаноле Р и кипятят с обратным холодильником в течение 30 мин. Затем через холодильник прибавляют 2,0 мл раствора бора фторида в метаноле Р и кипятят еще 30 мин, после чего прибавляют через холодильник 4,0 мл гептана Р и кипятят 5 мин. Охлаждают, прибавляют 10,0 мл раствора натрия хлорида насыщенного Р, встряхивают в течение 15 си прибавляют такой объем раствора натрия хлорида насыщенного Р, чтобы верхний слой поднялся к горлу колбы. Отбирают 2,0 мл верхнего слоя, помещают в делительную воронку, промывают тремя порциями воды Р, по 2 мл каждая, и высушивают над натрия сульфатом безводным Р.
Растворы сравнения, хроматографическая методика и оценка хроматограмм. Если в частной статье нет других указаний, поступают как указано в методе А.