

1.2. Основные понятия и законы, используемые в теории горения и взрыва
Химическая термодинамика – наука, которая изучает превращения различных видов энергии в химических реакциях (в том числе и при горении), процессах растворения, испарения, кристаллизации, адсорбции и др. Химическая термодинамика использует положения и выводы общей термодинамики, которая базируется на четырех основных законах, именуемых нулевым, первым, вторым и третьим законами (началами) термодинамики. Эти законы являются результатом обобщения человеческого опыта, практики. Данные, получаемые методами термодинамического исследования, являются исключительно достоверными, поскольку они опираются на основные законы природы о сохранении и преобразовании энергии.
Применительно к химическим процессам первый закон термодинамики имеет формулировку закона Гесса (закон постоянства сумм теплот реакций): суммарный тепловой эффект химической реакции не зависит от промежуточных состояний и пути перехода, а зависит только от начального и конечного состояний системы при условии, что температура и объем (давление) в начале и конце реакции одинаковы. Этот закон был установлен русским химиком акад. Г. И. Гессом в 1840 г.
Тепловым эффектом химической реакции называется теплота, которая выделяется или поглощается в ходе химической реакции. Стандартным тепловым эффектом химической реакции называется теплота, которая выделяется или поглощается в ходе химической реакции при стандартных условиях (температура 25 0С, давление 101325 Па). Единица измерения теплоты – Дж/моль или кДж/моль. Все химические процессы можно разделить на две группы: экзотермические и эндотермические.
Экзотермические – это реакции, при которых происходит выделение теплоты в окружающую среду. При этом запас внутренней энергии исходных веществ (U1) больше, чем образующихся продуктов (U2). Следовательно, ∆U0, а это приводит к образованию термодинамически устойчивых веществ.
Эндотермические – это реакции, при которых происходит поглощение теплоты из окружающей среды. При этом запас внутренней энергии исходных веществ (U1) меньше, чем образующихся продуктов (U2). Следовательно, ∆U>0, а это приводит к образованию термодинамически неустойчивых веществ.
В зависимости от условий протекания процесса, различают изохорный и изобарный тепловые эффекты.
Изохорным (QV) тепловым эффектом называют количество теплоты, которое выделяется или поглощается в ходе данного процесса при постоянном объеме (V = const) и равенстве температур конечного и начального состояния (Т1 = Т2).
Изобарным (Qр) тепловым эффектом называют количество теплоты, которое выделяется или поглощается в ходе данного процесса при постоянном давлении (р = const) и равенстве температур конечного и начального состояния (Т1 = Т2).
Для жидких и твердых систем изменение объема мало и можно принять, что Qр QV. Для газообразных систем
Qр = QV – ∆nRТ, (1.1)
где ∆n – изменение числа молей газообразных участников реакции
∆n = nпрод. реакции – nисх. веществ. (1.2)
Во всех случаях преобразование части внутренней (химической) энергии в тепловую (или другие виды) и наоборот, тепловой в химическую, происходит в строгом соответствии с законом сохранения энергии и первым законом термодинамики.
Теплотой образования данного соединения называется количество выделяющейся или поглотившейся теплоты при образовании 1 моля его из простых веществ в кДж. Теплоты образования простых веществ, находящихся при стандартных условиях в устойчивом состоянии, принимают равными нулю. Например, в реакциях
К(тв) + 1/2Сl(г) = КС1(тв) + 442,13 кДж
С(тв) + 1/2Н2(г) + 1/2N(г) = HCN(г) – 125,60 кДж
тепловые эффекты 442,13 кДж и -125,60 кДж представляют собой теплоты образования соответственно КСl и HCN.
Теплоты разложения указанных соединений на простые вещества согласно закону сохранения энергии равны по абсолютной величине, но противоположны по знаку, т. е. для КСl теплота разложения равна -442,13 кДж, а для HCN она составляет +125,60 кДж.
Теплотой сгорания называется теплота, выделяющаяся при полном сгорании 1 моля вещества в токе кислорода.
Величины тепловых эффектов химических реакций зависят от многих факторов: от природы реагирующих веществ, агрегатного состояния начальных и конечных веществ, условий проведения реакции (температуры, давления, объема систем, концентрации).
Тепловые эффекты многих химических и физических процессов определяют опытным путем (калориметрия) или рассчитывают теоретически, используя величины теплот образования (разложения) и теплот сгорания тех или иных химических соединений.
Согласно второму закону термодинамики, любая форма энергии может полностью преобразовываться в теплоту, но теплота преобразуется в другие виды энергии лишь частично. Условно запас внутренней энергии системы можно представить в виде двух слагаемых:
U = F + ТS, (1.3)
где F – полезная часть внутренней энергии, которая способна произвести работу, и которая, по предложению Гельмгольца, названа свободной энергией. В зависимости от условий протекания процесса свободную энергию можно представить в виде энергии Гельмгольца или энергии Гиббса.
Энергия Гельмгольца – это максимальная работа, которую может совершить система при равновесном проведении процесса при постоянных объеме и температуре, т. е. в изохорно-изотермических условиях (FV,T = U – ТS).
Энергия Гиббса – это максимальная работа, которую может совершить система при равновесном проведении процесса при постоянных давлении и температуре, т. е. в изобарно-изотермических условиях (Gp,T = Н – ТS).
ТS – непроизводительная часть, так называемая связанная энергия, которая ни при каких условиях не может быть превращена в полезную работу, и которая способна переходить только в теплоту и рассеиваться.
Энтропия (S) – это термодинамическая функция состояния, которая служит мерой неупорядоченности (беспорядка) состояния системы.
Свободная энергия в любой системе заключена в виде потенциальной энергии. По мере совершения системой работы ее энергия убывает. Чем больше система содержит свободной энергии, тем большую работу она может совершить. Так, более разряженный газ содержит меньше свободной энергии и больше связанной, чем сжатый газ при той же температуре. Следовательно, сжатый газ способен совершить больше полезной работы.
В ходе химической реакции свободная энергия уменьшается и при данных условиях достигает минимального значения, а система становится термодинамически устойчивой. Это соответствует состоянию равновесия. При изменении внешних условий равновесие тотчас же сместится, а в системе самопроизвольно возникнут процессы, которые вновь приведут свободную энергию к минимальному для данных условий уровню. Следовательно, состояние системы, соответствующее минимуму свободной энергии, является состоянием устойчивого равновесия при данных условиях. Из вышеизложенного можно сделать вывод: в изолированных системах самопроизвольно могут протекать только процессы, направленные в сторону понижения свободной энергии системы – это принцип минимума свободной энергии.
Энергия Гиббса (Гельмгольца) служит критерием самопроизвольного протекания химической реакции при изобарно-изотермических (изохорно-изотермических) процессах. Химическая реакция принципиально возможна, если G (F) 0 – это уравнение является условием возможности самопроизвольного течения реакции в прямом направлении. Химическая реакция не может протекать самопроизвольно, если свободная энергия возрастает: G (F) 0 – это условие невозможности самопроизвольного течения реакции в прямом направлении, но служит термодинамическим условием возможности самопроизвольного протекания обратной реакции. Если G (F) = 0, то реакция может протекать как в прямом, так и в обратном направлениях, т. е. реакция обратима.
Химическая кинетика – это учение о скорости химических реакций и ее зависимости от различных факторов: природы и концентрации реагирующих веществ, давления, температуры, катализаторов. О скорости химической реакции судят по изменению концентрации реагирующих веществ в единицу времени. Причем, с какой скоростью расходуются исходные вещества, с той же скоростью и накапливаются продукты реакции. Поэтому расчет можно проводить как по изменению концентрации исходных веществ, так и по изменению концентрации продуктов реакции.
Различают два вида скоростей: среднюю и истинную. Средняя скорость представляет собой отношения изменения концентрации к промежутку времени:
ср
![]() ,
(1.4)
,
(1.4)
где С1 и С2 – концентрации вещества в начальный (1) и конечный (2) момент времени.
Истинная скорость – это скорость химической реакции в данный момент времени, представляет собой первую производную концентрации по времени:
=
![]() .
(1.5)
.
(1.5)
Скорость всегда положительна, знак ± относится к изменению концентрации, если концентрация в ходе процесса увеличивается, то знак «+», а если уменьшается, то знак «−».
За скоростью процесса можно следить по изменению концентрации хотя бы одного из реагентов, учитывая его стехиометрический коэффициент. Для реакции вида
аA + вB → сC + dD
![]() .
(1.6)
.
(1.6)
Например, для реакции
СН4 + 2Н2О = СО2+4Н2
![]() .
.
Как видно, концентрация Н2О изменяется в 2 раза, а концентрация Н2 – в 4 раза быстрее, чем концентрации СН4 и СО2.
Зависимость скорости химических реакций от концентрации устанавливает закон действующих (действия) масс, сформулированный Гульдбергом и Вааге (1867), этот закон и является основным законом кинетики: скорость химической реакции при постоянной температуре пропорциональна произведению концентраций реагирующих веществ, взятых в степени стехиометрического коэффициента данного вещества в уравнении реакции.
Для гомогенной реакции общего вида
аA + вB → сC + dD
пр = k1CАа CВв, (1.7)
обр = k2Cсс CDd, (1.8)
где пр – скорость прямой реакции;
обр – скорость обратной реакции;
СА,CB,CC и CD – концентрации реагирующих веществ, например, моль/дм3;
а, в, с, и d – стехиометрические коэффициенты участников реакции;
k1 и k2 – коэффициенты пропорциональности, названные константой скорости химической реакции.
Для газов вместо концентрации можно использовать давления
пр = k1РАа РВв. (1.9)
Определим физический смысл константы скорости. Примем концентрации веществ А и В равными 1 моль/дм3 (СА = CB = 1 моль/дм3), тогда пр = k1. Таким образом, константа скорости – это скорость реакции, если концентрации реагирующих веществ равны 1 моль/дм3. Константа скорости зависит от тех же факторов, что и скорость, за исключением концентрации веществ. Причем, чем больше величина константы скорости, тем больше скорость процесса. Константу скорости можно найти опытным путем или рассчитать, используя уравнения, описывающие механизм химической реакции.
Зависимость константы скорости (и скорости) процесса от температуры определяет приближенное правило Вант-Гоффа: увеличение температуры на каждые 10 0С увеличивает скорость процесса в 2-4 раза.
Аналитическое выражение этого правила в общем виде:
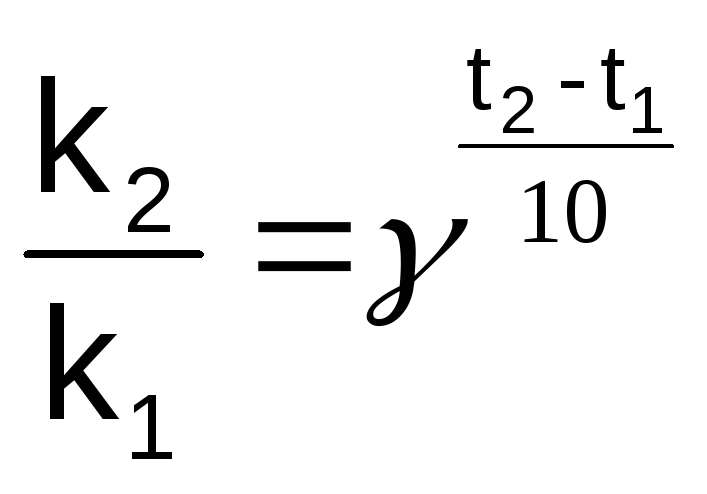 (1.10)
(1.10)
где k1 и k2 – константы скорости при температуре t1 и t2,
γ – температурный коэффициент Вант-Гоффа (γ = 2 4).
Правило Вант-Гоффа применяют при изменении температуры ниже 30 0С. При более высоких изменениях температуры используют уравнение Аррениуса.
lnk = В – А/Т, (1.11)
где А и В – постоянные величины для данной реакции.
Скорость любой химической реакции зависит от числа столкновений реагирующих молекул, так как число столкновения пропорционально концентрациям реагирующих веществ. Однако не все столкновения молекул приводят к акту их взаимодействия, а повышение температуры на 10 0С приводит к увеличению скорости в 2-4 раза, в то время как число столкновений увеличивается всего на 2 %.
Очевидно, что скорость реакции зависит не только от числа столкновений, но и от каких-то свойств сталкивающихся молекул. Это явление находит объяснение в теории активации С. Аррениуса (1889). Согласно этой теории, реакционноспособны только те молекулы, которые обладают запасом энергии, необходимым для осуществления той или иной реакции, т. е. избыточной энергией по сравнению со средней величиной энергии молекул. Такие молекулы получили название «активных» молекул. Переход неактивной молекулы в активную называется активацией. А избыточная энергия активной молекулы, благодаря которой становится возможной химическая реакция, носит название энергии активации (Ea). Единица измерения Ea Дж/моль или кДж/моль. Энергия активации бывает меньше энергии разрыва связей в молекуле, так как для того чтобы молекула прореагировала, вовсе не требуется полного разрыва связей, их достаточно лишь ослабить.
Величина энергии активации зависит от того, в какую реакцию эта молекула вступает. Иными словами, каждая химическая реакция характеризуется свойственной ей величиной энергии активации. Если реакция идет между атомами или свободными радикалами, то обычно она не превышает 40 кДж/моль, а если между молекулами, то – 150-250 кДж/моль, в полимерах она может достигать 400 кДж/моль. Если Ea выше наибольшей величины, то такая реакция невозможна.
Энергия активации молекул может быть снижена под воздействием внешних факторов: повышение температуры, лучистой энергии, катализаторов и др. Энергия активации проявляется в активных молекулах по-разному: активные молекулы могут обладать большей скоростью движения, повышенной энергией колебания атомов в молекуле и др. Следовательно, скорость химической реакции зависит от величины энергии активации: чем она больше, тем медленнее будет протекать данная реакция. С другой стороны, чем меньше энергетический барьер реакции, тем большее число молекул будет обладать необходимой избыточной энергией, и тем быстрее будет протекать эта реакция. Итак, скорость химической реакции в конечном итоге зависит от соотношения между числом активных и неактивных молекул. В теории активных соударений Аррениус показал, что количество активных молекул может быть вычислено по закону Максвелла-Больцмана:
Nакт = Nобщexp–Eа/RT, (1.12)
где Nакт – число активных молекул;
Nобщ – общее число молекул;
exp – основание натуральных логарифмов;
Eа – энергия активации, моль/Дж;
R – молярная газовая постоянная, Дж/(мольК);
T – абсолютная термодинамическая температура, К.
Уравнению (1.12) можно придать вид:
k = kоe–Eа/RT (1.13)
или после логарифмирования:
lnk
= lnkо
–
![]() ,
(1.14)
,
(1.14)
где k – константа скорости при обычных условиях;
kо – константа скорости при условии, что все столкновения приводят к реакции.
Сравнение уравнений (1.11) и (1.14) позволяет выяснить физический смысл констант в уравнении Аррениуса. A = Eа/RT характеризует энергию активации процессов (Eа), а B = lnkо, т. е. равно логарифму числа столкновений за 1 секунду в единице объема.
Если известны константы скорости k1 и k2 при двух температурах T1 и T2, то можно найти значение Eа из уравнения (1.14):
![]() (1.5)
(1.5)
или, заменяя натуральный логарифм десятичным, можно записать:
![]() .
(1.6)
.
(1.6)
Таким образом, рост скорости реакции с повышением температуры объясняется тем, что с увеличением температуры увеличивается не только средняя кинетическая энергия молекул, но и одновременно, как следует из уравнения (1.11), резко возрастает доля молекул, обладающих энергией выше определенного уровня, т. е. доля активных молекул, способных к реакции.
Скорость химической реакции можно регулировать с помощью катализатора. Вещества, которые участвуют в реакциях и изменяют (чаще всего увеличивают) ее скорость, оставаясь к концу реакции в первоначальном виде и количестве, называются катализаторами. Само изменение скорости химической реакции в присутствии катализаторов получило название катализа. По агрегатному состоянию каталитические реакции делят на гомогенные и гетерогенные.
Если от добавления катализатора к реагирующей смеси скорость реакции увеличивается, катализ называют положительным, если же реакция замедляется, то катализ называют отрицательными, а катализатор ингибитором. Ингибиторы применяют при тушении пожаров, например, хладоны.
Катализаторами могут быть самые разнообразные вещества в любом из трех агрегатных состояний: кислоты, соли, основания, оксиды, металлы, их атомы, молекулы или ионы, различные органические и органоминеральные соединения, газообразные вещества. В ряде случаев каталитическое действие оказывают всевозможные примеси (например, пыль), поверхность стенок сосуда, а также продукты реакции (в этом случае реакция называется автокаталитическая).
Катализ идет за счет перераспределения химических связей или сил электростатического взаимодействия участников реакции, механизм действия катализаторов связан с уменьшением энергии активации реакции за счет образования промежуточных соединений или образования активированного комплекса, при этом скорость процесса увеличивается в 2-3 раза.
Участие катализатора в реакции не отражается на стехиометрических коэффициентах и величине теплового эффекта химической реакции, а по сравнению с массой реагирующих веществ масса катализатора мала, но к концу реакции он выделяется в неизменном виде. Катализаторы действуют избирательно, т. е. скорость одних реакций данный катализатор ускоряет, а скорость других не изменяет или замедляет. Катализатор в одинаковой мере действует как на скорость прямой, так и обратной реакции. Он ускоряет лишь наступление состояния химического равновесия.
В ряде случаев присутствие посторонних веществ изменяет действие катализаторов: те вещества, которые усиливают положительную активность катализаторов, сами по себе являясь неактивными, называются промоторами или активаторами; те вещества, которые замедляют или практически полностью подавляют действие катализатора, называются каталитическими ядами; существуют вещества, присутствие которых не влияет на активность катализаторов (нейтральные).
Скорость гетерогенных реакций имеет ряд особенностей. Гетерогенные реакции идут на поверхности раздела фаз, которая и служит реакционным пространством (поверхностью). Поэтому первой особенностью кинетики этих реакций является влияние площади реакционной поверхности на скорость реакции. Вместо концентрации твердой фазы в уравнение вводят площадь поверхности этого вещества (S):
пр = k1CАа SВ. (1.17)
Второй особенностью гетерогенных процессов является зависимость скорости реакции от скорости подвода реагента в зону химической реакции, т. е. от скорости диффузии реагентов. Скорость диффузии увеличивают перемешиванием реакционной среды.
Скорость гетерогенной обратимой реакции определяется разностью скоростей прямой и обратной реакции (реакции = пр - обр) – это третья особенность гетерогенных процессов.
Все кинетические реакции различают по молекулярности и по порядку реакции. Молекулярность реакции определяется числом молекул, участвующих в элементарном акте химического взаимодействия. По этому признаку реакции разделяют на мономолекулярные, бимолекулярные и тримолекулярные – применяют для описания простых процессов.
Сложные реакции классифицируют по порядку, т. е. по сумме стехиометрических коэффициентов при концентрации реагирующих веществ. В простейшем случае порядок и молекулярность совпадают. Например, для реакции окисления NO до NO2 порядок и молекулярность совпадают и равны трем. Если = k 1СNO2СO2, то порядок равен n = 2 + 1 = 3. В сложных процессах порядок и молекулярность не совпадают, поскольку стехиометрическое уравнение описывает процесс в целом и не отражает истинного механизма реакции, протекающей через определенные стадии. Сложные реакции описываются кинетическим уравнением, содержащим несколько констант скоростей. К сложным реакциям относятся обратимые, параллельные, последовательные, сопряженные, цепные и другие реакции. Теория всех этих реакций основана на положении, что при протекании в системе нескольких реакций каждая из них проходит самостоятельно и к каждой из них применены уравнения кинетики простых реакций.
Параллельными реакциями называют реакции, которые имеют вид
А В + Д
С + К,
т. е. при которых одни и те же исходные вещества, одновременно реагируя, образуют разные реакции. Например, при разложении бертолетовой соли возможны процессы:
6КСlО3 2КСl + О2
3КСlО4 + КCl.
Последовательными называют реакции, которые протекают через ряд последовательных стадий по схеме:
А → В→ С→ D…
Примером служит гидролиз полисахаридов до моносахаридов.
Сопряженными называют реакции, протекающие по схеме:
А + В → М
А + С → N.
Реакция 1) может протекать самостоятельно, а реакция 2) только при наличии реакции 1). Например, йодоводород окисляется пероксидом водорода только при окислении сульфата железа пероксидом водорода. Побочный процесс называют химической индукцией, вещество А – актором, В – акцептором, С – индуктором.
Обратимыми называют реакции, скорость которых равна разности между прямой и обратной реакцией: реакции = пр - обр.
Например, образование сложного эфира.
Цепные реакции протекают путем образования цепи следующих друг за другом реакций, в которых участвуют активные частицы с насыщенными свободными валентностями – так называемыми свободными радикалами.
В целом скорость сложного процесса зависит от скорости самой медленной стадии, которую называют лимитирующей. Чаще всего, наблюдается закономерность: по какому механизму протекает самая медленная стадия, таков и механизм процесса в целом. Порядок реакции меняется от 0 до 3 и может быть как целым, так и дробным числом.
Таким образом, молекулярность характеризует элементарный механизм отдельных стадий сложного процесса, а порядок реакции характеризует формально-кинетическую зависимость скорости реакции от концентрации реагирующих веществ.