

Карцев В.Г. - Избранные методы синтеза и модификации гетероциклов. Том 1 (2003)(ru)
.pdfТаблица 3. Зависимость структуры конечного продукта от условий реакции метил-N-ацетилакрилата кислоты с алкилдиазоацетатом [34]
Раство- |
,D |
Соотно- |
Т, °С |
Выход образующихся структур, % |
|||
ритель |
раство- |
шение |
|
|
|
|
|
|
Пиразолин |
Циклопропан |
Цис : транс |
||||
|
рителя |
реагент. |
|
||||
|
|
17a |
18a, b |
18a : 18b |
|||
|
|
|
|
||||
|
|
|
|
|
|
|
|
СС14 |
0 |
2 : 1 |
22 |
98 |
– |
|
– |
СН2С12 |
1.60 |
1 : 1 |
22 |
30 |
– |
|
– |
|
|
2 : 1 |
22 |
60 |
30 |
1 |
: 3 |
|
|
2 : 1 |
40 |
30 |
68 |
1 |
: 2.25 |
|
|
3 : 1 |
22 |
Полимер |
– |
|
– |
СНС13 |
1.87 |
2 : 1 |
22 |
26 |
70 |
1 |
: 1 |
Как было показано в работе [34], варьирование условий реакции (табл. 3) позволяет целенаправленно получать производные пиразолинов 17a, b или циклопропанов 18a, b. Это можно объяснить протеканием реакции через промежуточный цвиттер-ион, который в полярных растворителях склонен к элиминированию молекулы азота с образованием циклопропанов (путь b), а в неполярном растворителе, даже при повышенной температуре, единственным направлением является путь а, т.е. циклизация в пиразолин (схема 3). При более высокой температуре и при проведении реакции в хлороформе наблюдается уменьшение содержания цис-циклопропана, что связано, по-видимому, с изменением конформации промежуточного цвиттер-иона [35].
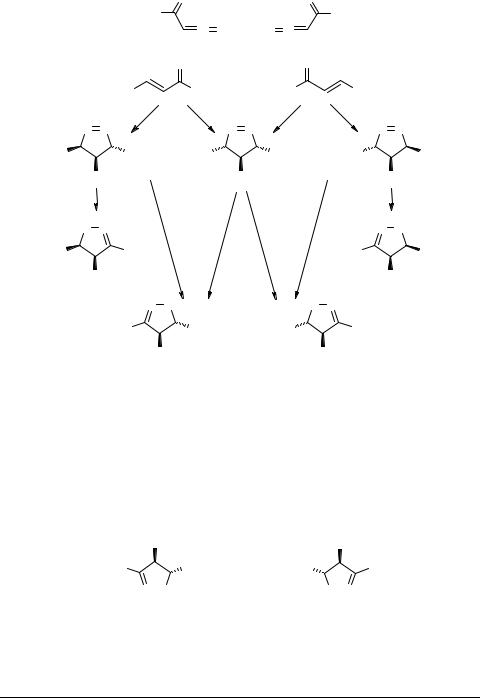
Наличие фенильного заместителя в β-положении эфира акриловой кислоты значительно дезактивирует С=С двойную связь за счет образования устойчивой системы сопряжения. По этой причине эфир коричной кислоты при нормальных условиях вступает в реакцию только с диазометаном и его алкильными производными (схема 3); при этом образуются соответствующие ∆-2 пиразолины 22а–d [44, 45]; использование фенилдиазометана требует увеличения времени реакции (соединение 23), а с алкилдиазоацетатами она протекает только при нагревании (350 ч, 52°С) [44] и приводит к изомерным пиразолинам 24а–с, 25 (табл. 4).
Первые работы [44, 45], связанные с изучением реакции диазосоединений с эфирами коричной кислоты, отмечали ее стереоспецифичность. Однако, более поздние попытки повторения этой реакции в аналогичных условиях при использовании стереооднородных Z- (или Е-) изомеров циннамата привели к образованию четырех изомерных пиразолинов 24с–e, 25 (или 24d–f, 25) [46, 47]. Вместе с тем, взаимодействие (Е-) этилового эфира коричной кислоты с метилдиазоацетатом, также как и (Е-) метилового эфира коричной кислоты с этилдиазоацетатом заканчивалось образованием смеси идентичных ∆-2 пиразолинов 24c–e и 24d–f. Оказалось, что каждый из промежуточно образовавшихся ∆-1 пиразолинов (син-, анти-) превращался в два изомерных ∆-2 пиразолина.
Избранные методы синтеза и модификации гетероциклов, том 1 |
11 |
|
|
|
|
|
|
|
Схема 4 |
|
MeO |
O |
|
|
O |
OEt |
|
|
+ |
− |
− |
+ |
|
||
|
|
N |
N |
N |
N |
|
|
|
|
+ |
|
|
+ |
|
|
|
|
O |
|
|
O |
|
|
|
Ph |
OEt |
|
MeO |
Ph |
|
|
|
N N |
|
N N |
|
|
N N |
|
MeO2C |
CO2Et |
MeO2C |
|
CO2Et |
MeO2C |
CO2Et |
|
|
Ph |
|
Ph |
|
|
Ph |
|
|
H |
|
|
|
|
H |
|
|
N N |
|
|
|
|
N N |
|
MeO2C |
CO2Et |
|
|
|
|
MeO2C |
CO2Et |
|
Ph |
|
|
|
|
Ph |
|
|
24c |
H |
|
|
H |
24f |
|
|
N N |
|
|
N N |
|
||
|
MeO2C |
CO2Et MeO2C |
CO2Et |
|
|||
|
Ph |
|
|
|
Ph |
|
|
|
24d |
|
|
|
24e |
|
|
Вместе с тем в реакции был зафиксирован аномальный ∆-2 пиразолин 25 (схема 5), что свидетельствовало о реализации двух путей присоединения диазосоединений к двойной связи: по правилу Ауверса (24c–f, структура А – главный продукт 90%), против правила Ауверса (24g, структура В – аномальный продукт).
Схема 5
|
Ph |
|
CO2Et |
||||
EtO2C |
|
|
CO2Me |
MeO2C |
|
|
Ph |
|
N |
|
N |
|
N |
|
N |
|
|
|
|
||||
|
|
|
H |
|
H |
||
|
А |
|
|
В |
|||
|
24c−f |
|
25 |
||||
главный продукт |
аномальный продукт |
||||||
12 |
Серия монографий InterBioScreen |

Таблица 4. Замещенные пиразолинкарбоновые кислоты, полученные взаимодействием ДС с производными коричной кислоты 22–24, 26*
|
R2 = Ar |
R4 |
R5 |
Выход, % |
|
R2 = Ar |
R4 |
R5 |
Выход, % |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
А |
В |
|
|
|
|
А |
В |
|
|
|
|
|
|
|
|
|
|
|
|
22a |
Ph |
H |
H |
100 |
– |
26a |
n-MeOPh |
H |
H |
100 |
– |
22b |
Ph |
Me |
H |
100 |
– |
26b |
n-MeOPh |
Me |
H |
100 |
– |
22c |
Ph |
Me |
Me |
100 |
– |
26c |
n-MeOPh |
Me |
Me |
100 |
– |
22d |
Ph |
H |
H |
100 |
– |
26d |
n-MeOPh |
Ph |
H |
63 |
37 |
23 |
Ph |
Ph |
H |
80 |
20 |
26e |
n-NO2Ph |
H |
H |
75 |
25 |
24a |
Ph |
H |
CO2Me |
95 |
– |
26f |
n-NO2Ph |
Me |
H |
66 |
34 |
24b |
Ph |
H |
CO2Bu |
98 |
– |
26g |
n-NO2Ph |
Me |
Me |
65 |
35 |
24c |
Ph |
H |
CO2Et |
85 |
– |
26h |
n-NO2Ph |
Ph |
H |
63 |
37 |
*Для соединений 22а R1 = Ph, R3 = H; для соединений 22b–d, 23–26 R1 = R3 = H
Вдальнейшем пиразолины со структурой В 23, 26d–h были зафиксированы и
вряде других случаев (табл. 4); увеличение их выхода наблюдалось при использовании фенилдиазометана (до 20%) и введении заместителей в п-положе- ние бензольного кольца циннамата (до 40%) [47, 48]. Конкурирующие направления взаимодействия ДС 2 (R4 = R5 = Ph) наблюдались и в случае его реакции с эфиром акриловой кислоты, где выход структуры В достигал 86%. Было отмечено, что образование такой структуры становится возможным при условии миграции фенильного радикала к атому азота [12].
Наличие в α-положении коричной кислоты N-ацетильного заместителя делает С=С кратную связь пространственно затрудненной. По этой причине ацетиламиноциннамат реагирует только с диазометаном и дает соответствующие пиразо-
лины 19a, b (СН2С12, 20°С, 70% [30]; СНС13 : (СН3)2О, 20°С, 90% [32]); с диазо-
уксусным эфиром он не взаимодействует даже в очень жестких условиях (кипячение в течение нескольких дней) [49]. Осуществить циклоприсоединение алкилдиазоацетатов к эфиру коричной кислоты и получить соответствующие ∆-2 пиразолины 21a, b удалось только в присутствии катализатора – тетраацетата диродия [31, 50]. При этом образуется исключительно ∆-2 структура В, в которой бензольное кольцо сопряжено со связью С=N пиразолинового цикла, что подтверждается данными 1Н ЯМР-, ИК- и УФ-спектроскопии [32].
Кроме реакции 1,3-диполярного циклоприсоединения ДС к активированным алкенам и образование производных пиразолинкарбоновой кислоты 27 было зафиксировано в реакциях циклизации гидразонов дикарбонильных соединений, сопровождающихся элиминированием уксусной кислоты [51] (схема 6).
Избранные методы синтеза и модификации гетероциклов, том 1 |
13 |
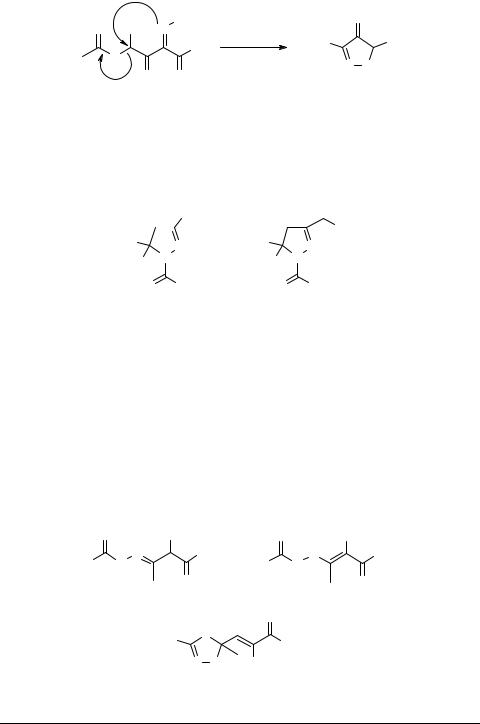
Схема 6
O Ph |
:N |
NH2 |
65°C |
|
O |
|
EtO2C |
Ph |
|||
|
|
OEt |
|||
O |
|
−AcOH |
|||
|
|
N N |
|||
O |
|
O |
|
|
|
|
|
|
H |
||
|
|
|
|
|
27 |
При взаимодействии дикарбонильных соединений с гидразинами [52] и гидразидами кислот [53, 54] наблюдается образование сложной смеси продуктов конденсации – гидразонов, енгидразинов и оксипиразолинов.
Схема 7
|
|
|
H (CO2Me) |
|
|
O |
||
HO |
|
|
HO |
|
|
|
OMe |
|
|
|
|
|
|
||||
|
|
|
|
|
|
|||
|
|
|
|
|||||
|
N |
N |
N |
N |
||||
t -Bu |
|
Ar |
||||||
|
|
|
|
|
|
|
||
|
O |
R |
|
O |
Ph |
|||
|
29a−f |
|
30 |
|
|
|
||
a R = H; b R = Me; c R = Et; d R = i-Pr;
e R = t-Bu; f R = Ar
Для соединений 29, 30 характерно существование равновесия между ациклической гидразоновой и циклической гидроксипиразолиновой формами [55, 56]. Содержание пиразолинов 29, 30 в смеси продуктов конденсации падает с увеличением объема заместителя в ацильной части молекулы и увеличивается при введении в дикарбонильную систему арильных заместителей с электроноакцепторными группировками.
Интересно отметить, что при конденсации 1,3-кетоэфиров с тиоацилгидразинами, наряду с открытыми формами 31а, b образуются 1,3,4-тиадиазолины 32
[57–60].
Схема 8
S |
R |
|
S |
H |
R |
Ph N N |
|
OMe |
|
OMe |
|
|
Ph N N |
||||
H |
|
O |
|
H |
O |
31a |
|
|
31b |
||
|
|
|
|
||
|
|
|
O |
|
|
|
Ph |
S |
OMe |
|
|
|
|
|
|||
|
|
|
|
||
|
|
N N |
R |
|
|
|
|
H |
|
|
|
|
|
32 |
|
|
|
14 |
Серия монографий InterBioScreen |
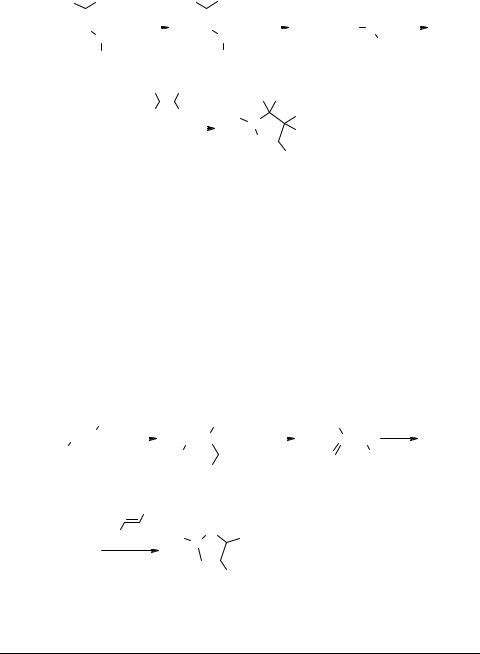
N-Замещенные пиразолинкарбоновые кислоты получены взаимодействием эфиров ненасыщенных кислот с N-фенилнитрилиминами [61–63].
Схема 9
O2N |
|
R4 |
O2N |
|
R4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
NaOH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N+ N |
|
|
|
||||
N NH |
|
|
|
|
|
|
|
|
N N |
|
|
|
|
|
|
|
|
R4 |
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
−H2O |
|
|
|
|
|
|
−NaNO2 |
|
|
|
|
|
||||||||||||||||
|
|
|
|
|||||||||||||||||||||||||
|
|
|
|
|
|
|
||||||||||||||||||||||
|
|
Ph |
|
|
|
|
|
Ph |
|
|
|
|
|
|
|
|
|
Ph |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
R3 |
|
|
CO2Me |
R1 CO2Me |
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
R2 |
|
|
R1 |
|
|
Ph |
N |
R2 |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
R3 |
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R4 |
|
|
|
|
|
|
|
R1 = R2 = R3 = H; R4 = Me [60]
R1 = R2 = R3 = H; R4 = Ph [61]
R1 = R2 = H; R3 = CO2Me; R4 = Ph [62]
R1 = R2 = Me; R3 = CO2Me; R4 = Ph [62]
R1 = R3 = H; R2 = CO2Me; R4 = Ph [12, 62]
R1 = H; R2 = OH; R3 = Me; R4 = Ph [62]
По данным авторов [64, 65] N,N-дизамещенные пиразолины могут быть получены при генерации дихлоркарбена межфазным методом в присутствии моноили 1,2-дизамещенных гидразинов. В этом случае первоначально образующийся илид 33 трансформируется в азометинимин 34, который вступает в реакцию 1,3-диполярного циклоприсоединения с диметилмалеатом, образуя пиразолин 35
(схема 10).
Схема 10
H |
|
R1 :CCl2 |
H |
+ |
|
R1 |
|
|
|
|
R1 |
+ |
|
|
|
||||||||
N |
|
N |
|
|
N |
|
|
NH |
|
|
|
|
|
|
N |
|
N |
|
|
||||
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
R |
|
H |
R |
|
|
|
|
|
|
|
|
Cl −HCl |
Cl |
|
|
|
|
R |
|||||
|
|
|
|
|
|
|
|
|
|
− |
|
|
|
|
|||||||||
|
|
|
|
|
|
Cl |
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
33 |
|
|
|
|
|
|
|
|
34 |
|
|
||||||
|
|
|
E |
|
|
|
|
R1 |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
E |
|
|
|
N |
|
E |
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
R N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
−HCl |
|
|
|
|
|
|
|
|
E |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
35 |
|
|
|
|
|
|
|
|
|
|
|
|
||
Избранные методы синтеза и модификации гетероциклов, том 1 |
15 |

Химические превращения пиразолинкарбоновых кислот
Пиразолины широко используются в синтетической практике как реагенты. Например, большое практическое значение имеет превращение эфиров пиразолинкарбоновых кислот 36 в пиразолы 37 под действием окислителей, воды [66] или при кислотном гидролизе [67]. Эти процессы протекают в мягких условиях и часто сопровождаются реакциями декарбоксилирования, дегалогенирования и миграцией алкильных или фенильных заместителей [68] (схема 11).
Схема 11
Ph |
|
H2O |
Ph |
||||
|
|
|
CO2Et |
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
N |
−CO2 |
N |
|
N |
|
|
|
|
|||||
|
|
|
|||||
|
|
H |
|
|
|
|
H |
36 |
|
|
37 |
||||
Щелочной гидролиз, как правило, приводит к раскрытию пиразолинового кольца и образованию различных карбонильных соединений [69].
Одним из наиболее изученных химических превращений пиразолинов является реакция их разложения c выделением азота, которая, как правило, сопровождается образованием соответствующих циклопропанов и ненасыщенных соединений. Эта реакция используется для получения производных циклопропана, в том числе веществ природного происхождения [70–74].
Термолиз эфира ∆-2 пиразолинкарбоновой кислоты 12а и его гомологов 12b, с завершается образованием только непредельных ациклических продуктов 39, в то время как их структурные ∆-1 аналоги 9a–e в аналогичных условиях дают смесь непредельных 39 и циклопропановых 38 производных [74, 75] (схема 12). Экспериментально установлено, что образованию алкенов способствует наличие нескольких алкильных заместителей в молекуле исходного пиразолина, а также асимметрично замещенного атома С3 [76–81].
При наличии двух карбоксильных групп в молекуле замещеного ∆-1 или ∆-2 пиразолина 3, 4, 6 разложение приводит к преимущественному образованию циклопропановых производных 38. Исключение представляет соединение 10а с геминальным расположением карбоксильных групп, которое разлагается с отщеплением одной из СООR, образуя исключительно алкены 39 [21, 79, 82]. Следует отметить, что и некоторые другие электроноакцепторные группы, такие как СN [19, 82–84], NO2 [85]) в гем-положении к СООR способствуют образованию алкенов. Обратное влияние оказывают электронодонорные группы (R2, R3 = Alk, Ph), при наличии которых реакции протекают в сторону образования циклопропанов [82].
Стереоспецифический характер процессов термического разложения ∆-1 пиразолинов был отмечен в работах [10, 21, 22, 86], где высказывалось предположение о протекании реакции через промежуточный цвиттерион.
16 |
Серия монографий InterBioScreen |
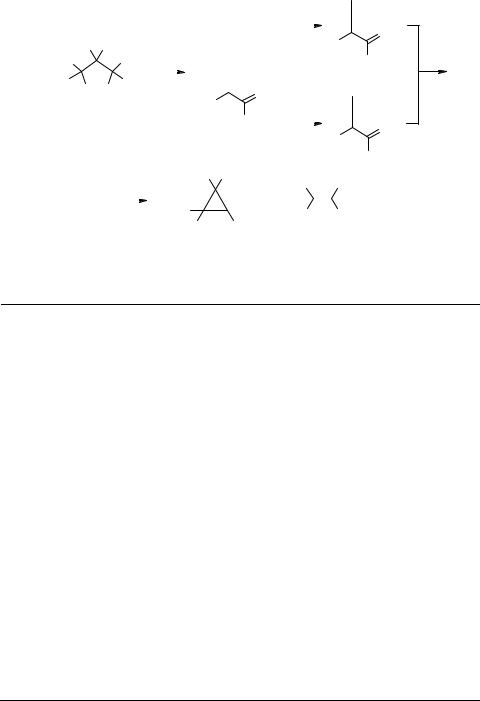
Схема 12
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH2 |
|||
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
R2 |
|
|
|
|
|
R3 |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
− O |
|||||
R2 |
|
R3 |
|
|
|
|
|
|
N |
|
|
−N2 |
R1 |
|
|||||||||||||||
|
|
|
R4 |
|
|
|
|
|
R5 |
|
|
|
|
|
|||||||||||||||
MeO2C |
|
|
R4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OMe |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
R1 N |
|
N R5 |
|
|
R2 |
|
|
|
|
|
R3 |
|
|
|
|
|
|
|
|
* |
CH2 |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
R1 |
|
|
− O |
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
3, 6, |
9−12 |
|
|
|
|
|
|
|
|
|
|
hν R2 |
|
|
|
|
R3 |
||||||||||||
|
|
|
|
|
|
|
OMe |
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
−N2 |
R1 |
* |
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
R2 |
R3 |
|
|
|
|
|
|
|
|
|
|
OMe |
||||||||||
|
|
|
|
|
|
|
R3 |
|
|
CO2Me |
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R4 + |
|
|
|||||||||||||
|
|
|
|
|
MeO2C |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
R2 |
|
|
R1 |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
R1 |
|
|
R5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
38 |
|
|
|
|
|
|
|
39 |
|
|
|
|
|
|
|
|
||||||||
Таблица 5. Соотношение продуктов 38, 39, образующихся при термолизе пиразо-
линов 3–6, 9, 12 [79]
Относительный и абсолютный выход продуктов термолиза, %
|
|
Циклопропан |
|
|
Алкен |
|
|
цис |
транс |
общ |
цис |
транс |
общ |
|
|
|
|
|
|
|
3a |
– |
95 |
95 |
– |
5 |
5 |
3b |
– |
95 |
95 |
– |
5 |
5 |
3c |
– |
95 |
95 |
– |
5 |
5 |
4а |
100 |
– |
100 |
– |
– |
– |
4b |
95 |
– |
95 |
5 |
– |
5 |
5d |
2 |
33 |
35 |
– |
65 |
65 |
6a |
– |
66 |
66 |
– |
– |
– |
6b |
– |
90 |
90 |
– |
10 |
10 |
9a |
– |
37 |
37 |
– |
63 |
63 |
9b |
23 |
– |
23 |
73 |
– |
73 |
9с |
– |
– |
– |
|
93 |
93 |
9d |
86 |
– |
86 |
14 |
|
14 |
9e |
11 |
72 |
83 |
|
17 |
17 |
10a |
– |
– |
– |
– |
– |
100 |
12a |
– |
– |
– |
– |
– |
100 |
12b |
– |
– |
– |
– |
– |
100 |
12c |
– |
– |
– |
– |
– |
100 |
12d |
50 |
– |
50 |
20 |
– |
20 |
|
|
|
|
|
|
|
Избранные методы синтеза и модификации гетероциклов, том 1 |
17 |
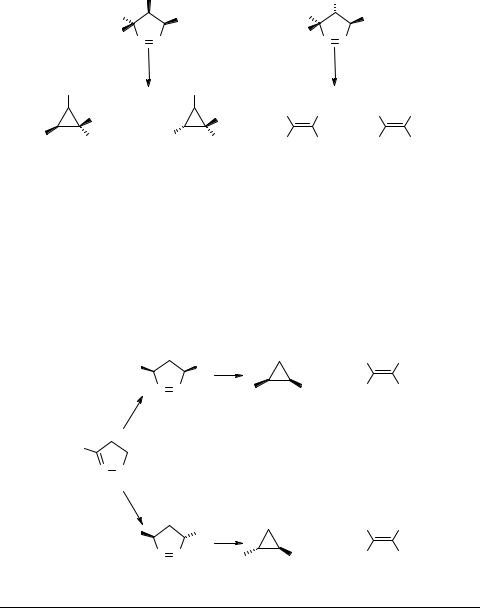
В более поздних исследованиях было обнаружено, что в ряде случаев разложение стереооднородных алкилзамещенных [15, 87] и фенилзамещенных [82, 88] ∆-1 пиразолинов протекает неспецифично, приводя к образованию смесей цис- и транс-изомерных циклопропанов 40а, b и алкенов 41а, b в различном соотношении (схема 13).
Схема 13
|
|
R2 |
|
|
R2 |
|
|
|
|
R1 |
|
|
R3 |
|
R1 |
R3 |
|
|
MeO2C N N |
|
MeO2C N N |
|
|
|||
|
R3 |
|
|
R3 |
|
|
|
|
|
CO2Me |
|
|
CO2Me |
R2 |
CO2Me |
H |
CO2Me |
R2 |
+ |
R2 |
+ |
+ |
|
|||
R1 |
R1 |
|
||||||
|
H |
R1 |
R2 |
R1 |
||||
|
40a |
|
|
40b |
|
41a |
|
41b |
Нарушение стереоспецифичности авторы [86, 89] связывают со способностью к миграции алкильных и фенильных групп.
∆-2 Пиразолины, согласно существующему мнению, при нагревании [89, 90] или под действием катализатора (200°С, КОН; [91–93]; Рt, EtОАс [94]) изомеризуются в соответствующие ∆-1 пиразолины 42, образующиеся в виде смеси син- (42а) и анти-изомеров (42b), разложение которых завершается образованием изомерных циклопропанов 43a, b и алкенов 44a, b. К подобному результату может приводить разложение ди- и тризамещенных пиразолинов (схема 14) [45].
Схема 14
MeO2C |
Me |
Me |
CO2Me |
+ |
|
||
N N |
Me |
|
|
CO2Me H |
H |
||
42a |
|
43a |
44a |
MeO2C 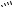 Me
Me
NN H
MeO2C |
Me |
H |
CO2Me |
+ |
|
||
N N |
Me |
|
|
CO2Me Me |
H |
||
42b |
|
43b |
44b |
18 |
Серия монографий InterBioScreen |

Преимущественное образование транс-изомера циклопропана 43b авторы
[95]связывают с его более высокой термодинамической стабильностью.
Вотличие от термолиза, фотолитическое разложение пиразолинов протекает по радикальному механизму [25, 38, 45, 47, 96, 97]. В ряде примеров использование фотолитического распада является более предпочтительным для получения циклопропанов, но, как правило, этот путь осложняется образованием 3–6 непредельных соединений.
Взаключение можно отметить, что простота получения и широкие возможности химической модификации пиразолинкарбоновых кислот делают этот класс соединений весьма полезным в органическом синтезе. На их основе легко получаются функционально замещенные карбо- и гетероциклические системы. Можно полагать, что дальнейшее изучение методов синтеза и реакционной способности пиразолинкарбоновых кислот позволит еще более расширить сферу их использования.
Литература
1.Мельников Н.Н., Новожилов К.В., Пылова Т.Н., Химические средства защиты растений, М.: Химия, 1985.
2.Общая органическая химия, под ред. Кочетковой Н.К., М.: Химия, 1985, т. 8,
с. 430.
3.Buchner E., Chem. Ber. 1888 21 2637.
4.Buchner E., Chem. Ber. 1890 23 701.
5.Buchner E., Papendieck A., Liebigs Ann. Chem. 1898 273 701.
6.Pechmann H., Burkard E., Chem. Ber. 1900 33 3590.
7.Buchner E., Chem. Ber. 1895 28 221.
8.Buchner E., Schroder H., Chem. Ber. 1902 35 782.
9.Auwers K., Ungemach G., Chem. Ber. 1933 66 1206.
10.Auwers K., Konig F., Chem. Ber. 1932 27 496.
11.Alphen J., Recl. Trav. Chim. Pays-Bas 1943 62 210.
12.Huisgen R., Seidel M., Wallbillig G., Tetrahedron Lett. 1962 17 3.
13.Huisgen R., Eberhar P., Tetrahedron Lett. 1971 45 4343.
14.Huisgen R., Angew. Chem. 1963 75 (16) 742.
15.Auken B., Rinehart K., J. Chem. Soc. 1962 84 3736.
16.Джемилев У.М., Докичев В.А., Султанов С.З., Изв. АН СССР, Сер. хим. 1989
(8) 1861.
17.Hassner A., Michelson M., J. Org. Chem. 1962 27 3974.
18.Eberhard P., Huisgen R., J. Am. Chem. Soc. 1972 94 (4) 1345.
19.Huisgen R., Stange H., Sturm H., Wagenhafer H., Angew. Chem. 1961 73 170.
20.Korobizina I.K., Rodina L.L., Methodikum Chimicum. 1974 6 105.
21.Young B., Lindenbaum S., J. Am. Chem. Soc. 1944 66 810.
22.Huisgen R., Angew. Chem. 1955 67 439.
23.Alphen J., Rec. Trav. Chim. Pays-Bas 1963 62 210.
24.Pechman H., Burhard E., Chem. Ber. 1900 33 3590.
25.Kohler H., Steele L., J. Am. Chem. Soc. 1919 41 1093.
26.Bregovec J., Jakovcic T., Monatsh. Chem. 1972 103 (1) 188.
Избранные методы синтеза и модификации гетероциклов, том 1 |
19 |

27.Suzuki M., Gooch E., Stammer C., Tetrahedron Lett. 1983 24 (36) 3839.
28.Mapelli C., Elrod L., Tetrahedron 1989 45 (14) 4377.
29.Wakamija T., Oda Y., Fujita H., Tetrahedron Lett. 1986 27 (19) 2143.
30.Cativiela C., Diaz M., Melendez E., Tetrahedron 1986 42 (2) 583.
31.Hiyama T., Kai M., Tetrahedron Lett. 1982 23 (20) 2103.
32.Анисимова Н.А., Дейко Л.И., Беркова Г.А., ЖОХ 1998 68 (7) 1165.
33.Анисимова Н.А., Дейко Л.И., Беркова Г.А., ЖОрХ 1991 28 (1) 205.
34.Анисимова Н.А., Дейко Л.И., Беркова Г.А., ЖОХ 1999 69 (9) 1529.
35.McGreen D.E., McKinly J., Can. J. Chem. 1971 49 105.
36.Stammer C., Tetrahedron 1990 46 (7) 2231.
37.Elrod L., Holt E., Stammer C., J. Chem. Soc. Chem. Commun. 1988 4 252.
38.Fernandez D., Frutos P., Marco L., Tetrahedron Lett. 1989 30 (23) 3101.
39.Yamonoi K., Ohfune Y., Tetrahedron Lett. 1988 29 (10) 1181.
40.Shimamoto K., Ohfune Y., Tetrahedron Lett. 1989 30 (29) 3803.
41.Dubois G.E., Crosby G.A., VcGarrayh G.V., J. Org. Chem. 1982 47 3270.
42.Pellicciari R., Natohini B., Marinozzi M., Tetrahedron Lett. 1990 31 (1) 139.
43.Shimamoto K., Ohfune Y., Tetrahedron Lett. 1989 30 (29) 3803.
44.Auwers K., Cauer E., Liebigs Ann. Chem. 1929 470 284.
45.Jones W., J. Am. Chem. Soc. 1955 77 6026.
46.Eberhard P., Huisgen R., Tetrahedron Lett. 1971 45 4337.
47.Bastiede I., Rousseav O., Pascot L., Tetrahedron 1974 30 3355.
48.Eberhard P., Huisgen R., J. Am. Chem. Soc. 1972 94 (4) 1345.
49.Дьяконов И.А., Алифатические диазосоединения, Ленинград: ЛГУ, 1978.
50.Анисимова Н.А., Дейко Л.И., Беркова Г.А., ЖОрХ 1998 34 (8) 1263.
51.Bertlo A., Hussel H., Liebigs Ann. Chem. 1927 457 278.
52.Singh S.P., Kumar D., Barta H., Naithani R., Can. J. Chem. 2000 78 (8) 1109.
53.Якимович С.И., Зерова И.В., Зеленин К.Н., в сб. Современные проблемы органической химии: Межвуз. сб. научн. тр. СПбУ, СПб, 1998, т. 12, с. 206.
54.Якимович С.И., Зерова И.В., ЖOрХ 1991 27 (5) 956.
55.Якимович С.И., Зерова И.В., Николаев В.Н., ЖOрХ 1986 22 (2) 286.
56.Якимович С.И., Николаев В.Н., ЖOрХ 1981 17 (2) 284.
57.Николаев В.Н., Якимович С.И., Зерова И.В., ХГС 1983 8 1044.
58.Bock H., Solouki B., Angew. Chem. Engl. 1981 20 427.
59.Kindermann, Kowski K., Muchall H., Rademacher P., Chem. Ber. 1993 126 2675.
60.Якимович С.И., Зерова И.В., Николаев В.Н., ЖOрХ 1983 19 (9) 1875.
61.Criegell R., Maschel A., Chem. Ber. 1959 92 2181.
62.Huisgen R., Angew. Chem. 1963 75 (13) 55.
63.Смирнов-Самков И.В., Успехи химии 1952 83 869.
64.Никирова Т.Ю., в сб. Современные проблемы органической химии: Межвуз. сб. научн. тр. СПбУ, СПб, 1998, с. 98.
65.Новиков М.С., Хлебников А.Ф., Костиков Р.Р., Изв. РАН, Сер. хим. 1996 (6) 1489.
66.Перекалин В.В., Липина Э.С., Сопова А.С., Непредельные нитросоединения, Ленинград: Химия, 1982, c. 430.
67.Parham W.Е., Bleasdale I., J. Am. Chem. Soc. 1950 72 (9) 3843.
68.Parham W.Е., Brakton H.G., J. Org. Chem. 1961 26 (6) 1805.
69.Parham W.Е., Bleasdale I., J. Am. Chem. Soc. 1966 88 3662.
20 |
Серия монографий InterBioScreen |