

9. Acidity and basicity |
409 |
Experimental and theoretical studies of the basicity and acidity of benzene-substituted indoles were performed169. In aqueous solution, the pKa values are above 4 for the bases and above 15 for the acids. Gas-phase ionization enthalpies have been calculated using the AM1 semiempirical method.
Politzer and coworkers170 have recently presented a linear relationship (r D 0.99) between the measured aqueous pKa values171 of a series of azines and azoles (9 pKa units) and the magnitude of the lowest value (Is,min,eV) of the average local ionization energy, I(r), on the molecular surface; I(r) is defined within the framework of SCF-MO theory as
I r D [ i r jεij/ r ] 33
where i r is the electron density of the ith molecular orbital at the point r, εi is the orbital energy and r is the total electron density. The correlation is reported in Figure 7.
VIII. NITRO DERIVATIVES
Kinetic and equilibrium acidities of several families of nitroalkanes have been discussed extensively in the chapter by Lewis172, where the effects of changing substituents and the nature of the base, together with the role of the solvent on rates of ionization and equilibria, have been considered.
Nonetheless, in the last decade nitroalkanes have been studied extensively because they are regarded as a good model for testing the applicability of new approaches to interpret proton transfer reactions. In fact, although nitroalkanes are strong carbon acids, their proton transfer behaviour is anomalous: for example, they are ‘slow’ acids (deprotonation of nitroalkanes and reprotonation of nitronate anions are slower than for other carbon acid families) and substituent effects within the series XC6H4CH2NO2 are greater on the rates of deprotonation than on the equilibrium acidities. Among the many approaches173 177, Bernasconi has formulated the Principle of Nonperfect Synchronization (PNS)178, according to which a product stabilizing factor (resonance or solvation) lowers the intrinsic reaction rate (k0) if it develops late, relative to the transfer of the negative charge from the base to the carbon acid, but enhances k0 if it develops early. The late development of solvation relative to the development of the nitronate anion at the transition state has been believed to be the main reason for the increase in the intrinsic rate constants (k0) with the increase in the DMSO content of the solvent. Essentially the same reason has been invoked to explain the variation of the disparity of Bronsted coefficients (‘imbalance’) in nitroalkanes with the solvent. Such imbalances are understood as the difference between the variation of the deprotonation rates with the pKa of the carbon acid (measured by ˛CH) and with the pKa of the base (measured by ˇB), ˛CH ˇB, assuming that ˇ is a rough measure of bond formation or charge transfer at the transition state.
On the same subject, Arnaut179 has recently published a paper in which the Intersecting State Model (ISM) proposed by Formosinho180,181 is used to provide a general approach to the deprotonation of carbon acids, including nitroalkanes. This model assumes that the activation energy of a reaction can be adequately calculated from the energetic variation occurring in the bonds of the reactants and products that suffer major geometry and/or frequency changes in the course of the reaction.
A. Nitromethane Derivatives
Since the equilibrium acidities of nitro-activated carbon acids have been considered172 in the preceding volume of this series, we show in Table 16 some significant values determined in DMSO from the enlarged compilation of DMSO equilibrium acidities proposed by Bordwell6b.

410 |
Silvia Bradamante |
|
|
TABLE 16. pKa values of some nitro-activated |
|
|
carbon acids in DMSO at 25 °C |
|
|
5-Nitrobarbituric acid |
0.8 |
|
PhSO2CH2NO2 |
7.1 |
|
CH2DCHCH2NO2 |
7.7 |
|
PhCH2NO2 |
12.3 |
|
Nitrocycloheptane |
15.8 |
|
Nitrocyclopentane |
16.0 |
|
CH3CH2NO2a |
16.7 |
|
Me2CHNO2 |
16.9 |
|
CH3NO2b |
17.2 |
|
Nitrocyclobutane |
17.8 |
|
Nitrocyclohexane |
17.9 |
|
p-NO2C6H4CH3 |
20.4 |
a Reference 182;
bpKa of nitromethane has been determined also in NMP (N-methylpyrrolidine-2-one) and found to be 19.8183.
TABLE 17. pKa values for some nitro-activated carbon acids in water and DMSO
|
H2O |
DMSO |
CH3NO2 |
10.2 |
17.2 |
PhCH2NO2 |
6.88 |
12.3 |
CH3CH(NO2)2 |
5.2 |
6.6 |
4-NO2C6H4CH2CN |
13.4 |
12.3 |
The differences in pKa values determined in water and DMSO, as shown in Table 17, might be interpreted as indicating differences in the solvation of the respective carbanions: the increase in pKa going from water to DMSO may be seen as the consequence of the loss of hydrogen-bonding solvation of the nitronate. The carbanion containing an aromatic ring seems to be better solvated in dipolar aprotic solvent, therefore its pKa decreases. A detailed study by Bernasconi184 and Bunnell indicates that pKa changes can be better rationalized by considering the solvent effect on the stability of the carbanion, the carbon acid and the hydronium ion. Although the pKa values of nitromethane and phenylnitromethane increase similarly, in nitromethane there is a strong destabilization of the carbanion, while in phenylnitromethane the destabilization of the carbanion is weaker but compensated by a greater stabilization of the carbon acid.
It has been found that a good correlation exists between Bordwell’s pKa values and heats of deprotonation ( HD) determined in DMSO7. In particular, when nitro compounds were considered together with ketones, they generated the following good (r D 0.993) linear correlation:
Hi° D 1.015 Gi° 4.43 |
34 |
Table 18 shows the free energies and enthalpies of ionization of some nitroalkanes.
The BDEs in the series NO2CXH2 (X D H, F, OH, NH2, CH3, CHDO and CHDCH2) have been calculated186 (MP4/6-31GŁŁ//HF/3-21G), and compared with those previously calculated187 and the experimental values188. The BDE were analyzed in terms of IBE (Intrinsic Bond Energies) and radical stabilization energies by comparing the BDE of C H and C NO2 in a series of related compounds. They are almost constant (59 62 kcal mol 1) due to a near cancellation of the increase in IBE (more positive)

|
9. Acidity and basicity |
411 |
||
TABLE 18. Thermodynamic properties for ionization of nitroalkanes in DMSO |
||||
at 25 °C |
|
|
|
|
|
H°a |
G°b |
S° |
|
|
i |
i |
i |
|
|
(kcal mol 1) |
(kcal mol 1) |
(cal mol 1 deg 1) |
|
Nitromethane |
20.1 š 0.9 |
25.1 |
16.8 |
|
Nitroethane |
18.3 š 0.9 |
24.4 |
20.5 |
|
Nitropropane |
19.1 š 0.9 |
24.8 |
19.1 |
|
2-Nitropropane |
19.3 š 0.9 |
24.7 |
18.1 |
|
Nitrobutane |
18.8 š 0.8 |
24.5 |
19.1 |
|
Nitrocyclobutane |
19.6 š 0.9 |
26.0 |
21.5 |
|
a H°i have been calculated using the formula H°i D HKDMSYLD C 48.0 š0.8 where the
value of 48.0 |
(š0.8) kcal mol 1 is the heat of autoprotolysis of DMSO185. |
b Gi° values |
have been calculated from the formula Gi° D 2.303 RT pKa C log 14 with |
SD D 0.1 kcal mol 1.
due to the substituent and an increase in radical stabilization energy (more negative) with the exception of NO2CHDO (52 kcal mol 1) and NO2CHDCH2 (71 kcal mol 1) where differences in radical stabilization have been invoked.
B. Structure of Nitroalkanes and their Conjugated Bases
Various studies189 at the semiempirical and ab initio levels have indicated that the nitromethyl anion adopts a planar C2v conformation, and the diand trinitromethyl anions have been predicted to be non-planar, the latter having a propeller-like geometry190,191. None of the basis sets used so far included both polarization and diffuse functions. Recently, an ab initio calculation (HF/6-31CCGŁŁ basis set, fully optimized geometry, ZPV (Zero Point Vibrational) energy differences included) has been performed192, indicating that the barrier to rotation around the C N bond is 128 kJ mol 1. The calculated EA of CH2NO2ž was 168 kJ mol 1, considerably different from the experimen-
tal 50 kJ mol 1. There was better agreement between the calculated proton affinity at C[PA(C) D 1579 kJ mol 1] and the experimental193 one (1500 kJ mol 1).
The electronic structures of CH(NO2)3 and its conjugated base have been recently studied at the HF/6-31GŁ and HF/6-31CCGŁŁ levels of theory by Cioslowski and coworkers194. The observed changes in geometry, the electron densities at the critical points and the GAPT (Generalized Atomic Polar Tensors)195 charges have revealed the presense of Y-aromaticity in C(NO2)3 anion. The computed geometry of the anion has indicated that the carbon atoms are coplanar with the nitrogens, while the oxygens are rotated out of plane leading to a structure with a D3 symmetry, as reported in Figure 8. This is in contrast to previous calculations (STO-3G level) by Edgecombe and Boyd190,191 in favour of the propeller structure with C3 symmetry. Absolute gas-phase acidities calculated with the different basis sets are shown in Table 19.
TABLE 19. Calculated absolute gas-phase acidites of CH(NO2)3
HF/6-31GŁ |
321 (kcal mol 1) |
HF/6-31 GŁŁ |
317.2a |
CC |
307.8b |
HF/6-31CCGŁŁ C ZPE |
|
exp |
n/a |
a At the HF/6-31GŁ geometries.
bZero-point energies calculated from unscaled vibrational frequencies at the HF/6-31GŁ level.
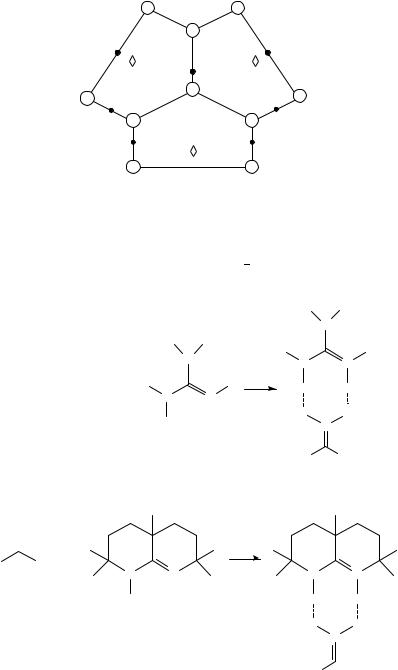
412 |
Silvia Bradamante |
O O
N
C
O 
 O
O
N N
O  O
O
FIGURE 8. Molecular graph of the C(NO2)3 anion. Heavy dots denote bond points and diamonds denote ring points. Reprinted with permission from Reference 194. Copyright (1991) American Chemical Society
Nitroalkanes are remarkably acidic in water (pKa of MeNO2 is 10.2172), but less in non-hydroxylic solvents (pKa of nitroalkanes are 28.6 30.4 in acetonitrile196). Evidence of their capacity to give tightly-bound ion pairs with bicyclic cations, as reported in the scheme, is provided on the basis of 1H NMR spectroscopy by Boyle and coworkers196.
|
|
|
|
|
|
X |
|
|
|
|
X |
|
+ |
|
|
|
|
N |
N |
|
|
|
|
|
|
||
R1R2 CH |
|
NO2 |
+ |
|
H |
H |
|
|
|||||
|
|
|
N |
|
N |
|
|
|
|
H |
|
O− |
+ O− |
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
R1 |
R2 |
In addition, it has been possible to isolate the complex 23 derived from the reaction of phenylnitromethane with the amidine 22.
Ph |
+ |
|
|
|
NO2 |
|
|
N + |
|
|
N |
N |
N |
|
|
H |
|
H |
H |
|
|
(22) |
O− |
O |
|
|
|
|
|
|
|
|
|
N + |
|
|
|
Ph |
|
|
|
|
|
(23) |

|
9. Acidity and basicity |
413 |
||
|
|
|
C(19) |
|
|
|
|
C(15) |
C(14) |
|
|
|
C(20) |
|
|
O(1) |
|
N(3) |
|
|
C(7) |
H(30) |
C(18) |
C(13) |
|
|
|||
C(2) |
N(1) |
H(31) |
C(8) |
C(12) |
|
|
|||
|
C(1) |
|
|
|
|
O(2) |
|
N(2) |
C(11) |
|
|
|
C(9) |
|
|
|
|
|
|
|
C(6) |
C(17) |
C(10) |
|
|
|
|
|
|
|
|
|
C(16) |
|
FIGURE 9. The X-ray crystal structure of complex 23, viewed from two different perspectives196. Reproduced by permission of the Royal Society of Chemistry
The X-ray structure of 23, shown in Figure 9, confirms that the hydrogen-bonding pattern and the internal geometry of the anion is similar to that reported for lithium phenylnitronate by Boche and coworkers197. The lithium phenylnitronate, prepared from phenylnitromethane and lithium ethoxide in ethanol, exhibits the presence of hydrogen bonds with ethanol incorporated into the crystal. In turn, this agrees with the conclusions derived for nitronates from acidity measurements on nitro compounds in protic solvents198 (Figure 10).
By using an ab initio MO approach at the HF/6-31GŁ level, Avakyan and Fateyev have determined structures and energies of the tautomeric forms of C- and N-nitro compounds MeCH2NO2 and MeNHNO2, and of their corresponding anions199. By comparing energy values (Etotal) of molecule pairs 24a 26a and 24b 26b, the true nitro compounds are found to be thermodynamically more stable than their aci-forms by 18.5 kcal mol 1 and 14 kcal mol 1, respectively. Among the aci-form conformers 26a and 26b, the Z,Z appeared to be the most stable using the AM1 approximation.
Although the equilibrium constant for the formation of aci-nitromethane (27) is very small, strongly basic solutions drive the equilibrium by formation of the aci-anion (28)200. When nitromethane was adsorbed on basic oxides (MgO and CaO) and zeolites (CsX), evidence for the formation of the aci-form was given by means of solid state NMR. The results indicate that nitromethane could be a much more discriminating probe than CO2 for studying basic sites in zeolites201.

414 |
|
|
|
|
Silvia |
|
|
|
|
|
|
|
|
|
|
|
|
13 |
|
|
|
|
|
|
|
|
|
|
|
|
|
12 |
|
|
22 |
|
23 |
|
|
|
|
|
|
O11 |
H11 |
|
21 |
|
|
|
|
4 |
5 |
|
H7 |
|
O30 |
26 |
25 |
24 |
|
|||
6 |
7 |
|
O9 |
|
|
N28 27 |
|
|
||||
|
|
|
|
|
H27 25 |
|||||||
|
N8 |
|
|
|
|
|
|
|||||
3 |
|
|
|
|
|
Li1 |
|
O29 |
H27 |
|
24 |
|
|
|
|
O10 |
|
|
O29 |
|
|
26 |
|||
|
|
|
|
|
O10 |
|
|
27 |
||||
|
|
|
|
|
O9 |
Li2 |
|
|
||||
|
1 |
|
5 |
|
|
|
|
23 |
||||
2 |
|
|
7 |
N8 |
|
|
N28 |
|||||
|
4 |
|
|
6 |
H31 |
|
31 |
O30 |
|
21 |
22 |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|||||
|
3 |
|
|
|
|
H7 |
|
32 |
|
|
|
|
|
|
|
|
|
|
33 |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||
|
|
2 |
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
FIGURE 10. Crystal structure of [PhCH NO2LiC ÐC2H5OH]n. The H atoms of the phenyl ring and the ethyl moiety have been omitted; the C atoms are only labeled by numbers197. Reproduced by permission of VCH, Weinheim
CH3XHNO2 |
! |
D |
|
! |
D |
|
CH3XD NO2 |
|
CH3X NOOH |
||
(24) |
|
(25) |
|
|
(26) |
(a) X D CH |
|
|
|
|
|
(b) X D N |
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
O |
CH3 |
O |
CH3 |
O |
CH3 |
||||
N X |
|
|
|
|
N |
X |
N |
X |
|
O |
|
H |
O |
|
O |
|
|||
H |
|
|
|
|
|
|
|
|
|
E,Z |
|
|
|
|
|
Z,Z |
|
E,E |
|
O |
|
|
|
|
H |
OH |
|
H |
O− |
H3 C N + |
|
|
|
|
|
N + |
|
|
N + |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|||
O− |
|
|
|
|
H |
O− |
|
H |
O− |
|
|
|
|
|
|
|
Base |
|
|
|
|
|
|
|
|
(27) |
|
|
(28) |
C. Nitroaryl-substituted Methanes
The various derivatives of o- and p-nitrotoluene are the most familiar class of compounds which can be considered vinylogous nitromethanes. Due to the low solubility in water of most of the compounds belonging to this class, pK values have been determined in aqueous-organic solvents. The availability of many pK scales in these solvent mixtures and the possibility of relating the relative acidities to the pKa scale has been discussed202. Some equilibrium constants are shown in Table 20.
While nitroalkanes are weaker acids in DMSO than in water, nitroarylmethanes show a decrease in their pKa values on increasing DMSO concentration.

|
9. Acidity and basicity |
|
415 |
||||||||||
TABLE 20. Equilibrium constants for nitroarylmethanes |
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Compound |
Solvent |
|
pK |
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Toluene derivatives |
|
|
|
|
|
|
|
|
|
|
|
|
|
4-Nitro |
5% water |
|
|
95% DMSO |
22.1 |
a |
|||||||
|
|||||||||||||
|
5% MeOH |
|
|
|
95% DMSO |
21.6 |
a |
||||||
|
|
|
|||||||||||
|
3% MeOH |
|
|
|
98% DMSO |
25.7 |
a |
||||||
|
|
|
|||||||||||
|
DMSO |
|
|
|
|
|
|
|
|
20.4 |
b |
||
2-Nitro |
water (estimate) |
|
25 |
a |
|||||||||
2,4-Dinitro |
40% water |
|
|
60% |
DMSO |
15 |
a |
||||||
|
|||||||||||||
|
30% water |
|
|
70% |
DMSO |
15.2 |
a |
||||||
|
|
||||||||||||
|
30% MeOH |
|
70% DMSO |
16.2 |
a |
||||||||
|
|
||||||||||||
|
40% MeOH |
|
60% DMSO |
16.6 |
a |
||||||||
|
|
||||||||||||
2,4,6-Trinitro |
MeOH |
|
|
|
|
|
|
|
|
15.6 |
a,a0 |
||
|
aqueous ethylenediamine |
14.45 |
c |
||||||||||
|
water |
|
|
|
|
|
|
|
|
13.61 |
d |
||
Diphenyl methane derivatives |
50% water |
|
|
50% |
DMSO |
10.5 |
e |
||||||
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4-Nitro |
50% water |
|
|
50% |
DMSO |
ca20 |
f |
||||||
|
|||||||||||||
4, 40 -Dinitro |
50% water |
|
|
50% |
DMSO |
14.94 |
e,g |
||||||
|
|||||||||||||
2, 4, 40 -Trinitro |
50% water |
|
|
50% |
DMSO |
12.19 |
d |
||||||
|
|||||||||||||
2, 20 , 4, 40 -Tetranitro |
water |
|
|
|
|
|
|
|
|
13.16 |
d |
||
2, 20 , 4, 40 , 6-Pentanitro |
50% water |
|
|
50% |
DMSO |
10.90 |
d,e |
||||||
|
|||||||||||||
50% water |
|
|
50% |
DMSO |
7.68 |
e |
|||||||
|
|||||||||||||
2, 20 , 4, 40 , 6, 60 -Hexanitro |
50% water |
|
|
50% |
DMSO |
5.01 |
e |
||||||
|
|||||||||||||
aReference 203. a0 Reference 204. bReference 205. cReference 206. dReference 207. eReference 208. fReference 209. gReference 210.
TABLE 21. Equilibrium acidity of nitro-substituted diphenylmethanes in 30% H2O 70% DMSO at 25 °C
|
|
|
n D number of NO2 |
|
pKa |
pKa |
|
|
|
|
0 |
30.6 |
(DMSO) |
||
|
|
|
|
27.2 |
(H2O |
|
DMSO) |
|
|
|
|
|
|||
|
|
|
|
28.9 |
(mean) |
|
|
4-Nitro |
|
1 |
16.90 |
|
12.4 |
||
4, 40 -Nitro |
2 |
15.54 |
|
1.36 |
|||
4, 40 |
, 400 -Nitro |
3 |
12.89 |
|
2.65 |
||
4, 40 |
, 400 |
, 2-Nitro |
4 |
9.93 |
|
2.96 |
|
4, 40 |
, 400 |
, 2, 20 -Nitro |
5 |
7.68 |
|
2.25 |
|
4, 40 |
, 400 |
, 2, 20 , 200 -Nitro |
6 |
7.20 |
|
0.48 |
|
4-Nitrotoluene is characterized by low acidity, while 2,4-dinitro- and 2,4,6- trinitrotoluene show higher acidities. Moreover, it is known that a p-nitro group has a higher resonance stabilizing effect in comparison with a o-nitro group, but the effect of additional substitution on the phenyl ring does not produce a proportional effect on the acidity because of the resonance saturation effect. Concerning the equilibrium acidity of diphenylmethane derivatives, the values shown in Table 21 indicate that it increases by increasing the number of nitro groups, but the resonance saturation effect appears to be

416 Silvia Bradamante
overcome by the effects produced by the absence of coplanarity of the two phenyl rings in the highly substituted nitroarylmethanes (n D 5 or 6). Those effects are mainly due to the conjugation of the sp2-hybridized carbanionic carbon with only one phenyl ring (the pycryl one in the case of n D 5). Accordingly, the pentaand hexasubstituted nitroarylmethanes behave as ˛-substituted 2,4,6-trinitrotoluenes. These compounds seem to be among the strongest benzylic-type carbon acids: they are more acidic than phenylnitromethane (pKa D 7.93 in 50% H2O 50% DMSO) and 2,4-dinitrophenylacetonitrile (pKa D 8.06 in 50% H2O 50% DMSO)211.
In the case of some nitrotriphenylmethanes, thermodynamic data determined in the H2O DMSO mixture212 indicate that (a) the increase in o-nitro groups in the three phenyl rings produces an important steric effect, (b) while the first p-nitro group produces an increase in acidity analogous to that found in diphenylmethane, the other p-nitro groups are less efficient: this can be ascribed to the propeller arrangement of the three phenyl ring in these anions. While the first p-nitro group could exert an additional stabilization of the negative charge, the other groups are less effective.
In the study of the reaction of 2,4,6-trinitrotoluene with base (NaOMe, under firstorder conditions), kinetic and equilibrium data for reaction in methanol DMSO have been obtained. Since it has been found that plots of logarithms of rate and equilibrium constants versus the mole fraction of DMSO are linear, the value in pure methanol have been extrapolated (Kp D 12.4 l mol 1)213.
IX. REFERENCES
1. J. W. Smith, in The Chemistry of the Amino Group (Ed. S. Patai), Wiley, London, 1968,
pp. 161 200.
2.D. K. Bohme, in Supplement F: The Chemistry of Amino, Nitroso and Nitro Compounds (Ed. S. Patai), Wiley, Chichester, 1982, pp. 731 762.
3.A. Albert and E. P. Serjeant, in The Determination of Ionization Constants. A Laboratory Manual, 3rd ed., Chapman and Hall, London, 1984.
4.(a) D. D. Perrin, Dissociation Constants of Organic Bases in Aqueous Solution, Butterworth, London, 1965.
(b) E. P. Serjeant and B. Dempsey, Ionization Constants of Organic Acids in Aqueous Solution, Pergamon, Oxford, 1979.
5.(a) R. A. Cox and K. Yates, Can. J. Chem., 61, 2225 (1983),
(b) A. Bagno, G. Scorrano, and R. A. More O’Ferrall, Rev. Chem. Intermed., 7, 313 (1987).
6.(a) F. G. Bordwell, Acc. Chem. Res., 21, 456 (1988).
(b) R. W. Taft and F. G. Bordwell, Acc. Chem. Res., 21, 463 (1988).
7.E. M. Arnett and K. G. Venkatasubramaniam, J. Org. Chem., 48, 1569 (1983).
8.F. Hibbert, Adv. Phys. Org. Chem., 22, 113 (1986).
9.E. N. Gur’yanova, I. P. Gol’dshtein and I. P. Romm, Donor Acceptor Bond, Wiley, New York, 1975.
10.W. B. Jensen, The Lewis Acid Base Concept. An Overview, Wiley, New York, 1980.
11.C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, 2nd ed., VCH, Weinheim, 1988.
12.(a) V. Gutmann, Coordination Chemistry in Non-Aqueous Solutions, Springer-Verlag, Vienna, 1968.
(b) V. Gutmann, Coord. Chem. Rev., 15, 207 (1975).
(c) V. Gutmann, The Donor Acceptor Approach to Molecular Interactions, Plenum, Press, New York, 1978.
13.P. -C. Maria, J. -F. Gal, L. Elegant´ and M. Azzaro, Thermochim. Acta, 115, 67 (1987).
14.(a) D. Gurka and R. W. Taft, J. Am. Chem. Soc., 91, 4794 (1969).
(b) R. W. Taft, D. Gurka, L. Joris, P. v. R. Schleyer and J. W. Rakshys, J. Am. Chem. Soc., 91, 4801 (1969).
15.(a) M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 98, 377 (1976).
(b) R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 98, 2886 (1976).

9. Acidity and basicity |
417 |
16.(a) R. W. Taft, J. -L. M. Abboud, M. J. Kamlet and M. H. Abraham, J. Solution Chem., 14, 153 (1985).
(b)M. H. Abraham, R. M. Doherty, M. J. Kamlet and R. W. Taft, Chem. Br., 22, 551 (1986).
(c)M. J. Kamlet, R. M. Doherty, J. -L. M. Abboud, M. H. Abraham and R. W. Taft, Chemtech, 566 (1986).
(d)M. J. Kamlet and R. W. Taft, Acta Chem. Scand., Ser. B, 39, 611 (1985).
17.(a) M. H. Abraham, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Faraday Trans. 1, 83, 2867 (1987).
(b)M. H. Abraham, P. P. Duce, P. L. Grellier, D. V. Prior, J. J. Morris and P. J. Taylor, Tetrahedron Lett., 29, 1587 (1988).
(c)M. H. Abraham, P. L. Grellier, D. V. Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 699 (1989).
(d)M. H. Abraham, P. L. Grellier, D. V. Prior, R. W. Taft, J. J. Morris, P. J. Taylor, C. Laurence, M. Berthelot, R. M. Doherty, M. J. Kamlet, J. -L. M. Abbound, K. Sraidi and G. Guiheneuf,´ J. Am. Chem. Soc., 110, 8534 (1988).
(e)M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris, P. J. Taylor, C. Laurence and M. Berthelot, Tetrahedron Lett., 30, 2571 (1989).
18.J. -L. M. Abboud, C. Roussel, E. Gentric, K. Sraidi, J. Lauransan, G. Guiheneuf,´ M. J. Kamlet and R. W. Taft, J. Org. Chem., 53, 1545 (1988) and preceding papers in the series.
19.M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris, P. Taylor, P. -C. Maria and J. -F. Gal,
J. Phys. Org. Chem., 2, 243 (1989).
20.(a) M. H. Abraham, Final Report, Scales of Hydrogen-Bonding Workshop, London, July 1 3, 1987.
(b)M. H. Abraham, G. J. Buist, P. L. Grellier, R. A. McGill, D. V. Prior, S. Olliver, E. Turner, J. J. Morris, P. J. Taylor, P. Nicolet, P. -C. Maria, J. -F. Gal, J. -L. M. Abboud, R. M. Doherty, M. J. Kamlet, W. J. Shuely and R. W. Taft, J. Phys. Org. Chem., 2, 540 (1989).
(c)M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 521 (1990).
21.J. Li, Y. Zhang, H. Ouyang and P. W. Carr, J. Am. Chem. Soc., 114, 9813 (1992).
22.J. E. Bartmess and R. T. Mclver, Jr., in Gas-Phase Ion Chemistry, Vol. 2 (Ed. M. T. Bowers), Chap. 11, Academic Press, New York, 1979.
23.J. Berkowitz, G. B. Ellison and D. Gutman, J. Phys. Chem., 98, 2744 (1994).
24.(a) S. G. Lias, J. F. Liebman and R. D. Levin. J. Phys. Chem. Ref. Data, 13, 695, (1984).
(b)S. G. Lias, Gas Phase Basicities and Proton Affinities. Additions and Corrections, 1987.
25.(a) M. Meot-Ner (Mautner) and L. W. Sieck, J. Am. Chem. Soc., 113, 4448 (1991).
(b)J. E. Szukijko and T. B. McMahon, Int. J. Mass Spectrom. Ion Processes, 109, 279 (1991).
(c)J. E. Szukijko and T. B. McMahon, J. Am. Chem. Soc., 115, 783 (1993).
26.D. A. Dixon and S. G. Lias, in Molecular Structure and Energetics, Vol. 2 (Eds. J. F. Liebman and A. Greenberg), VCH, New York, 1987, pp. 269 314.
27.D. D. Defrees and A. D. McLean, J. Comput. Chem., 7, 321 (1986).
28.(a) J. A. Pople, M. Head-Gordon, D. Fox, K. Raghavachari and L. A. Curtiss, J. Chem. Phys., 90, 5622 (1989).
(b)L. A. Curtiss, C. Jones, G. Trucks, K. Raghavachari and J. A. Pople, J. Chem. Phys., 93, 511 (1990).
(c)B. J. Smith and L. Radom, J. Am. Chem. Soc., 115, 4885 (1993).
29.A. Koppel, F. Anvia and R. W. Taft, J. Phys. Org. Chem., 7, 717 (1994).
30.(a) C. E. Doiron and T. B. McMahon, Can. J. Chem., 59, 2689 (1981).
(b)D. F. Drummond and T. B. McMahon, J. Phys. Chem., 85, 3746 (1981).
(c)S. M. Collyer and T. B. McMahon, J. Phys. Chem., 87, 909 (1983).
31.B. J. Smith and L. Radom, J. Am. Chem. Soc., 115, 4885 (1993).
32.J. Sunner and P. Kebarle, J. Am. Chem. Soc., 106, 6135 (1984).
33.(a) J. S. Uppal and R. H. Staley, J. Am. Chem. Soc., 104, 1238 (1982).
(b)J-F. Gal, R. W. Taft and R. T. Mclver, Jr., Spectros. Int. J., 3, 96 (1984).
34. (a) J. I. Brauman, J. A. Dodd and C. -C. Han, |
in Nucleophilicity (Eds. J. -M. Harris and |
S. P. McManus), Chap. 2, Advances in Chemistry |
Series, No. 215, ACS, Washington, D.C. 1987. |
(b)J. I. Brauman and C. -C. Han, J. Am. Chem. Soc., 110, 5611 (1988).
(c)T. B. McMahon and P. Kebarle, Can. J. Chem., 63, 3160 (1985).
(d)T. B. McMahon, T. Heinis, G. Nicol, J. K. Hovey and P. Kebarle, J. Am. Chem. Soc., 110, 7591 (1988).
418 |
Silvia Bradamante |
35.(a) J. W. Larson and T. B. McMahon, J. Am. Chem. Soc., 104, 5848 (1982).
(b)J. W. Larson and T. B. McMahon, J. Am. Chem. Soc., 105, 2944 (1983).
(c)J. W. Larson and T. B. McMahon, Inorg. Chem., 23, 2029 (1984).
(d)J. W. Larson and T. B. McMahon, J. Am. Chem. Soc., 106, 517 (1984).
(e)J. W. Larson and T. B. McMahon, J. Am. Chem. Soc., 109, 6230 (1987).
36.M. Meot-Ner (Mautner) and L. W. Sieck, J. Am. Chem. Soc., 108, 7525 (1986).
37.(a) E. P. Grimsrud, S. Chowdhury and P. Kebarle, Int. J. Mass Spectrom. Ion Processes, 68, 57 (1986).
(b)G. W. Dillow and P. Kebarle, J. Am. Chem. Soc., 110, 4877 (1988).
38.(a) J. W. Larson and T. B. McMahon, J. Am. Chem. Soc., 107, 766 (1985).
(b)J. W. Larson and T. B. McMahon, Inorg. Chem., 26, 4018 (1987).
(c)J. W. Larson, J. E. Szulejko and T. B. McMahon, J. Am. Chem. Soc., 110, 7604 (1988).
39.(a) J. W. Larson and T. B. McMahon, J. Phys. Chem., 88, 1083 (1984).
(b)J. W. Larson and T. B. McMahon, Can. J. Chem., 62, 675 (1984).
40.P. Kebarle and S. Chowdhury, Chem. Rev., 87, 513 (1987).
41.R. G. Keesee and A. W. Castleman, Jr., J. Phys. Chem. Ref. Data, 15, 1011 (1986).
42.M. Meot-Ner (Mautner), J. Am. Chem. Soc., 106, 1257 (1984).
43.(a) M. Meot-Ner (Mautner), J. Am. Chem. Soc., 105, 4912 (1983).
(b)M. Meot-Ner (Mautner) and C. A. Deakyne, J. Am. Chem. Soc., 107, 469 (1985).
(c)C. A. Deakyne and M. Meot-Ner (Mautner), J. Am. Chem. Soc., 107, 474 (1985).
44.M. J. Locke and R. T. Mclver, Jr., J. Am. Chem. Soc., 105, 4226 (1983).
45.(a) M. H. Abraham, J. Liszi and E. Kristof,´ Aust. J. Chem., 35, 1273 (1982): Calculations on Ionic Solvation, VII, and previous papers in this series.
(b)M. H. Abraham, E. Matteoli and J. Liszi, J. Chem. Soc., Faraday Trans. 1, 79, 2781 (1983).
46.J. Chandrasekhar, D. C. Spellmeyer and W. L. Jorgensen, J. Am. Chem. Soc., 106, 903 (1984).
47.(a) A. Pullman, in The World of Quantum Chemistry (Eds. B. Pullman and R. Parr), Reidel, Dordrecht, 1976, pp. 149 187.
(b)A. Pullman, in Quantum Theory of Chemical Reactions, Vol. 2 (Eds. R. Daudel, A. Pullman, L. Salem and A. Veillard), Reidel, Dordrecht, 1980, pp. 1 24.
(c)A. Pullman, Study Week on Chemical Events in the Atmosphere and Their Impact on Environment, November 7 11, 1983 (Ed. G B. Marino-Bettolo), Pontif. Acad. Sci. Scripta Varia, 56, 1985 pp. 25 50.
48.(a) D. H. Aue, H. M. Webb and M. T. Bowers, J. Am. Chem. Soc., 94, 4726 (1972).
(b)D. H. Aue, H. M. Webb and M. T. Bowers, J. Am. Chem. Soc., 98, 318 (1976).
49.R. W. Taft, in Proton-Transfer Reactions (Eds. E. F. Caldin and V. Gold), Chap. 2, Chapman and Hall, London, 1975.
50.R. W. Taft, in Kinetics of Ion Molecule Reactions (Ed. P. Ausloos), Plenum Press, New York, 1979, pp. 271 293.
51.R. W. Taft, J. -F. Gal, S. Geribaldi´ and P. -C. Maria, J. Am. Chem. Soc., 108, 861 (1986).
52.(a) M. Fujio, R. T. Mclver, Jr. and R. W. Taft, J. Am. Chem. Soc., 103, 4017 (1981).
(b)M. Mishima, R. T. Mclver, Jr., R. W. Taft, F. G. Bordwell and W. N. Olmstead, J. Am. Chem. Soc., 106, 2717 (1984).
53.M. J. Kamlet, R. M. Doherty, J. -L. M. Abboud, M. H. Abraham and R. W. Taft, Chemtech, 566 (1986).
54.T. Zeegers-Huyskens, J. Mol. Liq., 32, 191 (1986).
55.J. F. Gal and P. C. Maria, in Progress in Organic Chemistry, Vol. 17 (Ed. R. W. Taft), Wiley, New York, 1990, pp. 159 238.
56.V. Frenna, N. Vivona, G. Consiglio and D. Spinelli, J. Chem. Soc., Perkin Trans. 2, 1865 (1985).
57.A. D. Headley, J. Am. Chem. Soc., 109, 2347 (1987).
58.A. Beaten, W. De Proft, W. Langenaeker and P. Geerlings, J. Mol. Struct. Theochem, 306, 203 (1994)
59.A. D. Headley and M. E. McMurry, J. Phys. Org. Chem., 7, 63 (1994).
60.R. Barbucci, V. Barone, M. Micheloni and L. Rusconi, J. Phys. Chem., 85, 64 (1981).
61.D. N. Hague and A. D. Moreton, J. Chem. Soc., Perkin Trans. 2, 265 (1994).
62.M. Delfini, A. L. Segre, F. Conti, R. Barbucci, V. Barone and R. Ferruti, J. Chem. Soc., Perkin Trans., 900 (1980).
63.M. Micheloni, P. Paoletti and A. Bianchi, Inorg. Chem., 24, 3702 (1985).
64.A. Bencini, A. Bianchi, E. Garcia-Espana,˜ M. Giusti, M. Micheloni and P. Paoletti, Inorg. Chem., 26, 681 (1987).