

9. Acidity and basicity |
389 |
|
14.0 |
|
|
|
|
|
|
|
|
|
(15) |
|
|
|
|
|
|
|
|
|
|
|
|
|
12.0 |
|
|
|
|
|
|
|
|
|
|
|
10.0 |
|
|
Slope = 4.3 |
|
|
|
|
(14) |
||
|
|
|
|
|
|
|
|
||||
|
8.0 |
|
|
r = 0.989 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Slope = 2.7 |
||
−1 |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
r = 0.994 |
||
mol |
6.0 |
|
|
|
|
|
|
|
|
||
|
(16) |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|||
kcal |
|
|
|
|
|
|
|
|
|
||
4.0 |
|
|
|
|
|
|
|
|
|
|
|
G˚ (gas), |
2.0 |
|
|
|
|
|
|
(13) |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
0.0 |
|
(10) |
|
|
|
|
|
|
|
||
−δ∆ |
(17) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
(12) |
|
|
|
|
|
|
|
|
−2.0 |
|
(9) |
|
|
|
|
|
|
|
|
|
|
(8) |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
−4.0 |
(6) |
|
(7) |
(11) |
|
|
|
|
|
|
|
|
|
(5) |
|
|
|
|
|
|
|
|
|
−6.0 |
|
(4) |
|
|
|
|
|
|
|
|
|
|
(3) |
|
|
|
|
|
|
|
|
|
|
|
(2) |
|
|
|
|
|
|
|
|
|
|
−8.0 |
(1) |
|
|
|
|
|
|
|
|
|
|
|
−1.0 |
0.0 |
1.0 |
2.0 |
3.0 |
4.0 |
5.0 |
6.0 |
7.0 |
|
|
|
|
|
|
−δ∆G° (aq.), |
kcal mol−1 |
|
|
|||
FIGURE 3. |
Gas-phase vs |
aqueous basicities of substituted dimethylamines: (1) tert-amyl; (2) c- |
||
C6H11; (3) tert-butyl; (4) sec-butyl; (5) neoamyl; (6) isopropyl; (7) isobutyl; (8) n-propyl; |
(9) ethyl; |
|||
(10) |
methyl; |
(11) benzyl; |
(12) allyl; (13) propargyl; (14) CF3CH2; (15) NCCH2; |
(16) H; |
(17) |
(CH3)2NCH2. Reprinted with permission from Reference 57. Copyright (1987) American Chemical |
|||
Society
The gas-phase and solution basicities of various amines XCH2N(CH3)2 containing polar groups (X D CN, CCl3, C6F5, HC C, H2CDCH, C6H5) in different solvents have been compared59 in order to discover the factors that affect the ionization of difunctional molecules and to interpret structure acidity relationships correctly. The considered solvents were: H2O, MeOH, EtOH, 2-PrOH, EG (ethylene glycol), DMSO, TEP (triethylphosphate), AN (acetonitrile) and NB (nitrobenzene). As analysed using solvatochromic parameters ( Ł D the solvent’s dipolarity-polarizability constant, ˛ and ˇ D the solvent’s hydrogen-bond acidity and basicity constants, respectively), the solvation effects on basicities are not adequately accounted for by the solvent’s dipolarity-polarizability constant Ł . A better representation of the non-specific interactions between solvents and trimethylammonium ion is obtained using the product of Ł and the solvent dipole moment .
The basicity constants (Table 5) of the stepwise protonation of methylated tetramines (trienMe4 and trienMe6) have been considered and compared with those of the unsubstituted parent triethylenetetramine (trien).
R0 |
N(CH2)2NR(CH2)2NR(CH2)2NR0 |
R |
D |
R0 |
D |
H |
trien |
2 |
2 |
|
|
|
trienMe4 |
||
|
|
R D H, R0 D Me |
|||||
|
(3) |
R D R0 |
D Me |
trienMe6 |
|||
The log K values were found to decrease as the degree of methylation increased. Analysis of the data led to the conclusion that the first two protonations were on the terminal tertiary nitrogen, followed by the others on the inner nitrogens60.

390 Silvia Bradamante
TABLE 5. Basicity constants in water (ionic strength 0.1, at 25 °C) of the stepwise protonation of methylated tetramines
Amine |
Protonation |
log K |
trien Me6 |
I |
9.11 |
|
II |
8.35 |
|
III |
5.26 |
|
IV |
2.24 |
trienMe4 |
I |
9.19 |
|
II |
8.43 |
|
III |
5.62 |
triena |
IV |
2.73 |
I |
9.95 |
|
|
II |
9.31 |
|
III |
6.86 |
|
IV |
3.66 |
a Reference 62.
The protonation sequence of linear aliphatic polyamines H2N[CH2]k NH[CH2]l- (NH[CH2]m)nNH2 with k-m D 2 4, n D 0 3 can be determined using 13C NMR spectroscopy61.
The basicity constants (Table 6) of the stepwise protonation of polyazacycloalkanes have been determined.
H
N
HN |
NH |
N
( H )n
(4)
n = 4 |
N = 7 |
n = 5 |
N = 8 |
n = 6 |
N = 9 |
n = 7 |
N = 10 |
n = 8 |
N = 11 |
N = number of nitrogen atoms
TABLE 6. Basicity constants in water (in 0.5 M NaClO4, at 25 °C) of the stepwise protonation of polyazacycloalkanes
Protonation |
N D 7a |
N D 8a |
N D 9b,c |
N D 10b,d |
N D 11b,e |
N D 12b,e |
I |
9.83 |
10.01 |
9.59 |
9.85 |
9.79 |
9.75 |
II |
9.53 |
9.5 |
9.40 |
9.44 |
9.48 |
9.65 |
III |
8.84 |
9.1 |
8.77 |
8.95 |
9.02 |
8.88 |
IV |
6.72 |
8.29 |
8.27 |
8.56 |
8.64 |
8.96 |
V |
4.04 |
5.01 |
6.37 |
7.79 |
8.06 |
8.12 |
VI |
2.43 |
3.71 |
4.22 |
5.24 |
6.44 |
7.82 |
VII |
2.3 |
2.98 |
3.24 |
3.84 |
4.49 |
5.66 |
VIII |
|
1.97 |
2.31 |
3.02 |
3.58 |
4.27 |
IX |
|
|
1.8 |
1.97 |
2.76 |
3.58 |
X |
|
|
|
1.8 |
2.26 |
2.62 |
XI |
|
|
|
|
1.7 |
2.3 |
XII |
|
|
|
|
|
1.0 (estimated) |
a Reference 63.
b0.15 M NaClO4. c Reference 64.
dReference 65. eReference 66.

|
|
9. Acidity and basicity |
391 |
|||
|
|
|
|
|
|
O |
|
|
|
|
|
H |
H |
|
|
|
NH |
|
N |
N |
H |
|
NH |
HN |
|
|
|
|
|
|
|
|||
N |
H |
|
|
|
HN |
NH |
H |
|
|
|
|
|
|
N |
N |
NH |
|
HN |
|
|
H N |
N H |
|
|
|
O |
O |
NH |
NH |
NH |
|
|
||
N |
|
|
|
N |
N |
|
|
|
|
|
|||
H |
|
|
|
|
H |
H |
(5) |
|
|
(6) |
|
|
(7) |
|
|
H |
O |
H |
|
|
H |
|
N |
|
N |
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
H |
H |
|
|
|
|
|
N |
N |
NH2 |
H N |
|
N |
H |
|
|
|
|
|
|
H N |
N H |
N |
NH2 |
N |
|
N |
H N |
N H |
H |
|
H |
|
H |
|
N |
N |
|
|
|
|||
|
|
O |
|
|
H |
|
H |
|
|
|
|
||
|
|
|
|
|
|
|
(8) |
|
|
(9) |
|
|
(10) |
TABLE 7. Basicity constants in water (ionic strength D 0.1 M TsNa at 25 °C) of the stepwise protonation of compounds 5 10
Protonation |
5 |
6 |
7 |
8 |
9 |
10a |
I |
10.50 |
10.65 |
9.35 |
10.10 |
9.15 |
10.20 |
II |
10.20 |
10.55 |
9.25 |
10.10 |
9.00 |
9.25 |
III |
9.25 |
9.70 |
8.35 |
9.30 |
8.20 |
8.75 |
IV |
8.00 |
9.20 |
6.80 |
8.70 |
7.20 |
4.10 |
V |
7.05 |
8.20 |
5.65 |
7.70 |
3.70 |
³2 |
VI |
6.40 |
7.75 |
5.55 |
7.00 |
3.40 |
³1 |
VII |
|
6.85 |
|
|
|
|
VIII |
|
6.50 |
|
|
|
|
a Reference 68.
Macrocyclic polyamines have been extensively studied by Lehn’s group67 and the protonation constants of polyamines [24]ane-N6 (5), [32]ane-N8 (6) and [27]ane-N6O3
(7) have been evaluated and compared with those of the acyclic analog (8) and of the macrocycles [24]-N6O2 (9) and [18]-N6 (10)68 (Table 7).
Proton affinities of some ˛-amino acids have been presented by Bojensen69 who used the kinetic approach developed by Cooks and coworkers70. Alternatively, Gorman et al.71 used FT ion cyclotron resonance spectrometry to determine gas-phase basicities of 20 common ˛-amino acids, from which proton affinities have been derived. The relative ordering of PA of the two research groups presents noticeable differences relative to histidine, glutamine, glutamic acid, alanine, methionine and threonine. According to Gorman, discrepancies could arise from Cook’s method tested for monofunctional compounds, although this argument has been challenged by the Danish group72.
The solution basicities in water at 25 °C of ortho-, meta- and para-substituted primary, secondary and tertiary anilines have been widely discussed by Smith1. Preliminary gas-phase data were reported by Bohme2. Subsequently, the gas-phase basicities (proton
392 |
Silvia Bradamante |
affinities) of 15 substituted anilines were determined by measuring proton transfer equilibria by means of a pulsed electron beam high pressure mass spectrometer73. Nitrogen protonation occurs when - and -donating substituents are present at the para position and electron-donating substituents are on nitrogen. Conversely, -donating substituents at the meta position favour ring protonation: these anilines show higher basicities than expected. However, when a strong electron-withdrawing substituent, such as CN and CF3, is at the meta position, nitrogen protonation occurs again because ring protonation is not favoured. A linear relationship between the gas-phase basicities and the basicities in water of nitrogen-protonated anilines was obtained with an attenuation factor of ca 4.
B. Amidines and Guanidines
The general topic of the basicity of amidines has been recently covered in a chapter74 in the book The Chemistry of Amidines and Related Compounds. Here, the general scheme rationalizing the dependence of basicity on the nature of the substituents is outlined.
The basicity of compounds containing the amidino group NDC N depends on the substituents at the three sites (both nitrogens and the amidino carbon atom) and the protonation site is the imino nitrogen atom75. It has been shown76 that, at any of the three sites, in the series of monosubstituted amidines, their pKa values obey Hammett’s equation.
pKa D pKa0 |
19 |
In addition, the pKa values of amidines substituted at the imino nitrogen atom correlate with the pKa values of the corresponding primary amines and, in turn, this correlation can be used to predict the pKa values of trisubstituted amidines. The pKa values of a series of trisubstituted formamidines (C6H5 NDCH NR1R2), with variable substituents at the amino nitrogen atom, correlate with the pKa values of corresponding secondary amines R1R2NH; the correlations are in the following form:
pKa(amidine) D pKa0 C ˛[pKa(amine) pKa(aniline)] |
20 |
The pKa values of amidines containing two substituted phenyl rings (one at the imino nitrogen and the other at the amidino carbon atom) do not obey a simple dual parameter equation, but fit the following equation:
pKa D pKa0 im im F F im F |
21 |
where im and F are the Hammett-type constants at the imino nitrogen and the functional carbon, respectively, and represents the mutual interaction of the substituents.
Following the same reasoning, a general equation has been proposed to predict pKa values of amidines containing various substituents at the three sites of the amidino group:
pKa D pKa0 1 1 2 2 3 3 12 1 2 13 1 3 23 2 3 |
22 |
where k and l are the constants of the substituents at each pair of the site (k and l) of the amidino group, and k,l represents the mutual interaction between the substituents.
Molecular recognition by abiotic synthetic receptors has been considered an important goal in biorganic chemistry. The guanidinium group (pKa ca 13.5), which remains protonated over a wide range of pH, is able to form (strongly zwitterionic) hydrogen bonds with carboxylates and phosphates. This behaviour has been exploited to build optically active abiotic receptors77. Lehn’s group has shown that a bis-naphthoyl ester of the rigid bicyclic guanidine (11) can complex sodium p-nitrobenzoate, sodium p-methoxybenzoate

|
|
9. Acidity and basicity |
393 |
|
|
|
N |
Cl− |
|
|
|
|
|
|
|
H |
H |
|
|
|
N |
+ N |
|
CO2 − Na + |
|
H |
H |
|
|
O |
O |
O |
O |
|
+
NO2
(11)
|
N |
|
|
|
H |
|
|
|
H |
+ |
|
|
|
|
N |
N |
|
|
|
|
|
|
||
H |
|
H |
|
|
O |
|
|
O |
O |
O |
− |
O |
|
|
|
|
|||
O |
|
|
|
|
NO2
(12)
and sodium phenylacetate. The chirality of 11 forces any substrate to bind in a dissymmetric environment, allowing the enantioselective recognition of chiral carboxylic acids. When N-acetyl and N-tert-butoxycarbonyl derivatives of sodium tryptophan were examined, two diastereomeric complexes were detected.
The X-ray crystal structure of the nitrate salt of the chiral bicyclic guanidine78 (Figure 4) indicates that the counter ion is bound by two virtually parallel hydrogen bonds to both of the protonated guanidinium nitrogen atoms. The nitrate ion is not coplanar with the guanidinium system, but tilted by 20.8° with respect to the plane through the atoms N(1), N(2), N(3) and C(1). The extraordinary basicities79 of these bicyclic guanidines is well illustrated by the fact that, if an equimolar amount of the free base of 13 is added to an acetonitrile solution of the salt of acetyl-D,L-alanine and of 1,8-bis-(dimethylamino)naphthalene

394 |
|
Silvia |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
C(93) |
|
|
|
|
|
|
|
|
C(92) |
C(94) |
|
C(164) |
|
|
C(7) |
|
|
|
|
|
C(163) |
|
|
C(4) |
|
|
|
|
||
|
|
|
|
|
|
|
C(91) |
C(95) |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
C(165) |
C(20) |
C(6) |
N(1) |
|
C(3) |
C(13) |
|
|
|
|
|
|
|
C(9) |
|
|||
|
|
|
|
|
|
|
|
|
|
C(162) |
C(16) |
|
|
|
|
|
|
C(11) |
|
|
O(2) |
C(5) |
|
|
|
|
|
||
|
C(18) |
C(1) |
H(2) |
C(2) |
O(1) |
C(14) |
|||
|
C(15) |
|
|||||||
|
|
|
Si(1) |
||||||
|
C(161) |
|
|
|
|
|
|
|
|
|
|
|
H(5) |
N(3) |
|
|
|
|
|
|
C(19) |
Si(2) |
|
N(2) |
|
|
|
||
|
|
C(21) |
|
|
|
|
C(8) |
C(12) |
C(101) |
|
|
|
|
H(3N) |
|
H(2N) |
|||
|
|
|
|
|
|
|
|
C(10) |
|
|
|
C(17) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C(105) |
C(102) |
|
C(175) |
C(171) |
|
O(3) |
|
|
|
|
|
|
|
|
O(4) |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N(4) |
|
|
|
|
C(174) |
C(172) |
|
|
|
|
|
C(104) |
C(103) |
|
C(173) |
|
|
O(5) |
|
|
|
|
|
FIGURE 4. Molecular structure of nitric salt of chiral bicyclic guanidine (13)78. Reproduced by permission of the Royal Society of Chemistry
(proton sponge), 1H NMR shows evidence of the complete deprotonation of the proton sponge cation and the formation of diastereoisomeric host guest complex.
|
|
N |
|
|
H2 C |
N |
+ |
N |
CH2 |
|
|
|
||
But(Ph)2 Si O |
H |
|
H |
O Si(Ph)2 But |
(13)
Multiply aminated phosphinimines behave as exceptionally strong bases and this property has been recently exploited by Schwesinger in a variety of organic reactions80. In particular, among the group of kinetically highly active uncharged peralkylated polyaminophosphazenes, P4-t-Bu (14) is one of the most hindered and basic, more than 24 pK units stronger than 1,8-bis-(dimethylamino)naphthalene or triethylamine and ca 18 pK units stronger than DBU.
|
|
|
|
|
|
|
|
Me |
|
|
|
|
|
|
||||
|
|
|
|
Me |
|
|
|
|
|
|
|
Me |
||||||
|
|
|
|
|
|
|
|
|
|
|||||||||
Me2 N |
|
|
|
|
|
|
|
|
NMe2 |
|||||||||
|
N |
|
|
|||||||||||||||
Me2 N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
P |
|
N |
|
|
P |
|
N |
|
P |
|
NMe2 |
||||||
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NMe2 |
|||
Me2 N |
|
|
|
|
|
|
N |
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
Me2 N |
|
|
|
|
NMe2 |
||||||||||
|
|
|
P |
|
||||||||||||||
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
NMe2 |
||||||||
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
(14) |
|
|
|
|
|
|
|||||

9. Acidity and basicity |
395 |
C. Amidates and Amides
The scale of hydrogen-bond basicity pKHB, initially developed by Taft and coworkers14, has been extended by Abraham and coworkers81, and is based on the formation of hydrogen-bond complexes of a base B with a hydrogen-bond reference donor as 4- fluorophenol in CCl4 at 25 °C:
B |
C |
4-FC6H4OH |
! |
4-FC6H4OH |
Ð Ð Ð |
B |
(23) |
|
|
|
|
|
|
KHB D [4-FC6H4OH Ð Ð Ð B]/[B][4-FC6H4OH] and pKHB D log KHB
The pKHB values can be transformed to give the parameter ˇ2H D pKHB C 1.1 /4.636, which ranges from 0 (no basicity) to 1 (basicity of HMPA). This scale has been extended to many organic functionalities including amidines82 and amides83.
On the basis of IR evidence it has been proposed84 that alkylamidates are the strongest known hydrogen-bonding carbonyl bases and that the order of hydrogen-bond basicity may be alkylamidates ³ HMPA > vinylogous amides ³ benzamidates > amides (Table 8).
TABLE 8. Hydrogen-bond basicity of amidates RCON NC Me3 and, for comparison, some carboxyamides in CCl4
Amidates |
pKHB |
ˇ2H |
i |
3.32 |
0.95 |
R D Pr t |
||
Bu |
3.25 |
0.94 |
1-Adamantyl |
3.56 |
1.01 |
N,N-Dimethylformamide |
2.10a |
0.69 |
N,N-Diethylacetamide |
2.47a |
0.77 |
1-Methyl-2-piperidone |
2.60a |
0.80 |
N,N0 -Dimethyl-N,N0 -trimethyleneurea |
2.79a |
0.84 |
3-Dimethylamino-5,5-dimethylcyclohexenone |
2.92 |
0.87 |
HMPA |
3.56b |
1.00 |
a Reference 83. b Reference 14.
V. NITROGEN ACIDS
When substituted with identical groups, NH hydrogen acids are more acidic than CH carbon acids, and thus ammonia is more acidic than methane, and aniline more acidic than toluene, etc. Notwithstanding the great importance of metal amides as bases, systematic study of the acidity of NH nitrogen acids has received attention only since the development of acidity scales in organic solvents for investigating the acidity of CH carbon acids. This delay may be due to the fact that, with the exception of the sulphonamides, cyanamides and imides, the majority of nitrogen acids are weak. Moreover, carbanions are ubiquitous intermediates in organic chemistry and nitranions (the conjugate bases of nitrogen acids), which are usually harder species than carbanions, give rise to a complex landscape of interactions with the cation and solvent. Structural investigations of nitranion salts have appeared more recently than analogous studies of carbanionic and organometallic species. It will therefore be useful to describe some recent important results concerning the characterization of nitranions in terms of their bonding, structure and interactions with the cation and solvent.

396 |
Silvia Bradamante |
A. Structural Characterization of Metal Amides in the Solid State and in Solution
X-ray crystal studies have contributed much towards clarifying the molecular arrangements of metal amides in the solid state, while 13C, 15N and lithium countercation NMR have revealed the structural and dynamic aspects of various interactions in solution. Collum85 pioneered NMR spectroscopic, crystal structure and theoretical approaches to the awareness of this class of compounds. Structural studies in the solid state involved metal dialkylamides86, isopropylamides87, allylamides88, piperides86a,89, amidomagnesiates90, various silylamides91, cyanamide92 and chiral amides93. Dimers94, tetramers, and up to decameric and dodecameric95 oligomers, as well as adamantanoid96 arrangements, have been found. Computations97 have very often closely mimicked experimental results, and the role of structure upon reactivity has also been investigated (e.g. in carbonylation reactions98).
Boche and coworkers drew attention to the solid state structure of mixed amine metal amide99 and silylamide nitrile complexes100 as models of reaction intermediates.
Polyazacycloalkanes of the series [3k]ane-Nk (7 k 12) are known to form polynuclear complexes with metal ions such as Mn2C , Co2C , Ni2C, Zn2C and Cd2C , and can generally include more than one ion, thus forming dinuclear complexes; trinuclear complexes have been found with k D 12, 13. When Pd2C was used, diand trinuclear complexes with azamacrocycles [18]ane-N6 and [21]ane-N7 were found and their structures have been ascertained by means of X-ray analyses. In the case of the trinuclear complex, deprotonation of a secondary amino group occurs both in solution and the solid state101 (Figure 5).
|
|
C(8) |
|
N(4) |
C(7) |
|
|
N(5) |
|
C(6) |
|
C(5) |
C(4) |
C(9) |
|
Pd(3) |
|
|
N(3) |
Cl(3) |
|
|
C(10) |
C(3)
 N(6)
N(6)
Cl(1)
Pd(2)
Pd(1)  C(11)
C(11)
N(2)
Cl(2)
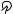 C(12)
C(12)
N(7)
C(2) |
N(1) |
C(14) 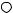 C(13)
C(13)
C(1)
FIGURE 5. ORTEP view of the complex cation [Pd3([21]aneN7 )Cl3 2C . The deprotonated nitrogen is N3101. Reprinted by permission of the Royal Society of Chemistry

9. Acidity and basicity |
397 |
N43 |
Li314 |
N33 |
|
||
O14 |
|
|
Li412 |
O44 |
O34 |
|
||
|
|
|
N13 |
|
|
Li123 |
|
|
O24 |
Li234 |
|
|
N23 |
|
FIGURE 6. ORTEP diagram of the lithium N-methylpseudoephedrate (I) solid state tetramer. Hydrogen atoms have been omitted for clarity. Reprinted with permission from Reference 102. Copyright (1990) American Chemical Society
Solid state investigations have been generally corroborated by solution studies. The X- ray structure of lithium N-methylpseudoephedrates102 indicates the tetrameric aggregation in which the lithium atoms are chelated between oxygen and nitrogen (Figure 6). The structure in solution has been demonstrated to be tetrameric in benzene by means of vapour pressure osmometry (VPO, 37 °C) and cryoscopy (5.5 °C).
Nitranion salts of heterocyclic NH-azoles have been investigated in the solid state103 and solutions of different solvents, and with various counterions104. The initial explorative 1H NMR study has been more recently extended to the 13C NMR investigation of nitranions PhN X, in which the nature of X allows the inclusion of amine, nitramide, sulphonamide, phosphonamide, cyanamide and carboxamide functionalities105. The results show the considerable structural reorganization undergone by the precursor nitrogen acid PhNHX upon deprotonation: the effect on 13C shifts exerted by the [2.2.1] cryptand indicates the few cases in which ion pairing and/or aggregation occurs. Correlative analysis of the NMR data shows the effect of the substituent in PhN X, revealing that charge transfer from the nitrogen atom to the phenyl ring is dominated by the resonance component of the effects exerted by the substituent X. Unlike the analogous acid base pair of toluene carbon acids PhCH2X and benzyl carbanions PhCH X, there is no correlation whatsoever between Cpara of PhN X and Bordwell’s acidities of PhNHX in DMSO. The energetics of the deprotonation process are dominated by the polar-inductive component of the effect exerted by the substituent X. The different sensitivities of charge release from nitrogen in nitranions, as well as of the acidities of nitrogen acids, are accounted for by structural arrangements in these species. The results indicate that the lone pair developing on the nitrogen atom upon deprotonation has a limited chance of providing an extra resonance stabilization to the anion in comparison with the neutral. The nitrogen atom adopts a planar sp2 configuration (the two electron pairs occupying a p and an sp2 orbital, respectively) when X is a group containing a system. If nitranions similarly adopt such a configuration at the nitrogen atom, with colinearity of the p orbitals of the phenyl ring,

398 |
Silvia Bradamante |
the nitrogen atom and the substituent X, the electron pair residing in the nitrogen sp2 orbital can enhance electron release from the nitrogen atom to both the phenyl and the substituent. The absence of any direct overlap of the nitrogen sp2 orbital with the p orbital of X explains why the acidity of nitrogen acids is only marginally affected by substituents characterized by strong resonance electron-withdrawing (EW) power.
These considerations have been confirmed106 by the finding that nitranion 15N resonances can undergo displacements of opposite sign in relation to the shift of the precursor nitrogen acid. Low-field and high-field displacements respectively occur when a sp2 or a p charge is generated. Thus, in the anion of 2- and 4-picoline, there is a substantial increase in the -negative charge on the pyridyl nitrogen: in these anions a high-field displacement of the 15N resonance is observed in relation to the neutral precursor. Deprotonation of pyrrole, as well as of aromatic amines and carboxamides, generates and locates the negative charge in an sp2 orbital of the nitrogen atom: the 15N shift of pyrrole nitranion is low-field displaced in relation to that of the neutral pyrrole. A more subtle interpretation of the low-field shift of the 15N nitranion resonance of aromatic or heteroaromatic amines comes from a detailed RHF/6-31GŁ study107 of pyridyl amine nitranions. The isotropic low-field shift of 15N aromatic amine nitranions is associated with a decrease in the charge density on nitrogen: in a simplified picture, the sp2 electron pair generated by the deprotonation of the NHacid ‘pushes away’ the p electron pair on nitrogen. Anionic species generated by the CH-deprotonation of 2- and 4-alkylpyridines should be considered real nitranions. In fact, it has been shown that metalation of 4-alkylpyridines originates in the solid state N-metalated-4-alkylidene-1,4- dihydropyridines108. Similar results have also been found in solutions of dipolar aprotic, metal-coordinating solvents (DMSO)109 and the results have been used to probe the EW capacities of substituents at the methylidene group in position 4.
EWG |
EWG |
EWG |
|
− |
|
N |
N |
N |
(15) |
|
− |
|
|
13C and 15N NMR studies of nitranions of the pyridyl type have been extended to a large variety of azine110 and azole109a,111 systems. The charge densities on the nitrogen atoms of these nitranions have been empirically computed using 15N shift/ -charge relationships: the resulting 13C and 15N charge maps are consistent and congruent.
Nitranions originated by the deprotonation of aldehyde phenylhydrazones can be seen as diazaallylic anions: -charge delocalization has also been investigated using the NMR approach in these systems112.
B. Amine Derivatives
Quantitative and semiquantitative equilibrium acidity scales have been established for weak carbon acids in cyclohexylamine (CHA)113, dimethoxyethane (DME)114, dimethylsulphoxide (DMSO)115 and THF116.
Fraser and coworkers116 measured the relative acidities of 15 weakly acidic hydrocarbons in THF using 13C NMR spectroscopy. However, as the experiments were