

22. Radical anions and cations derived from CDC, CDO or CDN groups 1311
synthetic utility of this chemistry (Scheme 30). Electrophiles such as cyclohexanone, benzaldehyde, diphenyldisulfide and carbon dioxide successfully reacted in yields of 65 80%. In sense, this technique is superior to the related cyclizations of the 5-hexenyl radical involving n-Bu3SnH in that there is no loss of functionality at the radical center after cyclization.
HO CH3
O
Sml2 |
CH3 |
86% |
(28) |
|
THF (t-BuOH) |
(>150:1) |
|||
|
|
HMPA
A similar cyclization of a ketyl anion (produced via reaction with Sml2) onto an allene was reported by Gillmann in 1993 (equation 29)85 The regiochemistry and high diasteroselectivity associated with this process was explained on the basis of a ‘chelation-controlled’ cyclization (e.g. TS 48). The same reduction carried out with n-Bu3SnH resulted in only a 37% yield and a 5 : 1 ratio of diastereomers.
O
CH3 O
• |
H |
O
O
Sml2
CH3 O
THF t-BuOH
HO
(29)
82% (15:1) O
+CH3 O
HO
Analogous intramolecular ‘chelation-controlled’ ketone/olefin couplings with Sml2, in which SmC3 was complexed in a cyclic manner to the ketyl anion and a ˇ-carbonyl of an ester or amide functionalilty, were reported as early as 1987 (Scheme 31)86. The cyclized samarium intermediate 49 could be further reacted with added electrophiles such as aldehydes or ketones87.
Cyclization of ketyl anions onto allyl sulfides or sulfones were reported to yield 2- vinylcyclopentananols according to equation 3088.
X
|
Sml2 |
|
|
THF/HMPA |
(30) |
CHO |
OH |
|
H |
|
|
|
|
X = SPh (50%)
X = SO2 Ph (80%)

1312 Daniel J. Berger and James M. Tanko
|
|
O− |
|
|
• |
Sm+3 |
|
|
|
H O |
|
|
|
OCH3 |
|
|
• |
|
|
|
(48) |
|
|
|
|
|
Sm+3 |
O |
O |
O− |
|
|
|
O |
|
R1 |
Y |
Sml2 |
|
|
R2 |
R1 |
Y |
|
|
|
R2 |
|
Sm+3 |
|
|
Sm+3 |
1 O− |
O |
|
1 O− |
O |
R |
|
Sm+2 |
R |
|
|
|
H2 C |
|
|
|
Y |
|
Y |
|
Sm(lll) |
|
|
||
R2 |
|
|
R2 |
|
|
|
|
||
(49) |
|
|
|
|
E + |
|
|
|
|
R1 OH O |
R1 OH O |
ECH2 |
CH3 |
Y |
Y |
R2 |
R2 |
|
SCHEME 31 |
(b) Intramolecular additions to other remote -systems. In 1986, Shono demonstrated the feasibility of intramolecular ketyl anion cyclizations onto an aromatic ring (equation 31)89. These reductions were achieved electrochemically in isopropanol, using a Sn electrode and Et4NC X electrolyte. Use of LiClO4 as an electrolyte yielded only the reduced product 51.
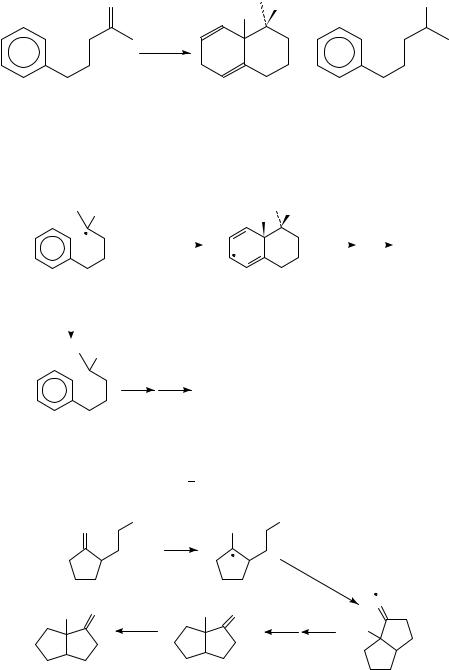
22. Radical anions and cations derived from CDC, CDO or CDN groups 1313
O |
OH |
|
OH |
|
H |
|
|
|
e− |
+ |
|
|
i-PrOH |
|
|
|
|
|
|
|
n-Bu4 NX |
|
|
|
Sn cathode |
(51) |
|
|
(50) |
|
|
|
70% |
7% |
(31) |
|
|
|
The effect of the counterion on the outcome of this reaction was attributed to ion-pairing effects (Scheme 32). It was suggested that the ketyl anion 52 is more easily reduced when LiC is the counterion, and that this second reduction occurs faster than cyclization90.
|
|
O−M+ |
|
O−M+ |
||||
|
|
|
|
H |
||||
|
|
|
|
|
|
|
|
50 |
|
|
|
|
|
|
|||
|
|
|
fast when |
|||||
|
|
|
M + = n-Bu4 N+ |
|||||
(52) |
|
|
|
|
|
|
||
e− |
|
fast when |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
M+ = Li+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O−M+
−
51
SCHEME 32
Intramolecular cyclizations of ketyl anions (generated electrochemically at a Sn cathode in isopropanol) onto a remote C N were reported in 199291. ˛-Hydroxyketones are produced in yields ranging from 60 80%, depending on the structure of the substrate (Scheme 33).
CN |
O− |
CN |
|
O |
|
||
|
e− |
|
|
OH O |
OH NH |
N |
|
−O |
|||
H + |
|
||
H2 O |
|
|
SCHEME 33

1314 |
Daniel J. Berger and James M. Tanko |
SmI2-promoted, |
intramolecular ketone/alkyne couplings were reported in 1990 |
(equation 32)92. Yields were low (30 50%) for unactivated alkynes (Y D H or CH3), but improved to 50 75% when Y D Si CH3 3, Ph or CO2Et. Also, yields were typically much better for ketones than for aldehydes.
|
|
|
|
|
|
|
Y |
Y |
|
O |
Sml2 |
|
|
||
|
|
|
|
|
(32) |
||
|
|
|
|
|
THF |
||
|
|
|
|
|
|
||
|
|
|
R |
HMPA |
R |
||
R = H or CH3 |
|
|
|
|
|
OH |
|
|
|
|
|
|
|
|
|
C |
X |
|
|
|
C |
X |
|
βC α |
C |
|
|
|
C |
C |
|
|
|
|
|||||
|
O_ |
|
|
|
|
|
O_ |
C X −C C
O
FIGURE 4. Resonance forms of ˛,ˇ-unsaturated carbonyl compounds
O O
CH2 OTs
O− |
|
O |
|
|
− |
|
CH2 OTs |
CH2 OTs |
|
|
SCHEME 34
22. Radical anions and cations derived from CDC, CDO or CDN groups 1315
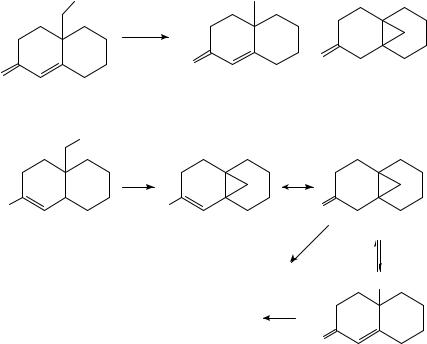
f. Radical anions generated from ˛,ˇ-unsaturated carbonyl compounds. The principles underlying the chemistry of radical anions generated from ˛,ˇ-unsaturated carbonyl compounds (mainly aldehydes, ketones and esters) are similar to those discussed earlier for >CDOž in general. The key difference is that, as a result of the transfer of spinand/or electron-density to the ˇ-carbon through resonance (Figure 4), new reaction pathways become accessible. There are examples in which the ˇ-carbon seems to be reacting both as a nucleophile and as a radical, several of which are discussed below.
(i) ‘Nucleophilic’ reactions of >CDC CDOž . In 1981, Gassman reported that electrochemical reductions of enones possessing a good leaving group at the υ position yield cyclopropanes in excellent (>80%) yields93. The mechanism suggested for this process is summarized in Scheme 34. (Similar reductions were achieved in the 1960s by Stork using Li/NH3, although in much lower yields.)94
In contrast, under conditions of photoinduced electron transfer, Givens and Atwater found that the major product of the reaction is C X reduced product 53 (equation 33)95. It was proposed that under these conditions, the cyclopropylcarbinyl radical 55 undergoes further (reversible) ring opening (55 ! 56, Scheme 35). The reversibility of this ring opening was demonstrated by generating radicals 55 and 56 independently via reaction of the corresponding alkyl bromides with n-Bu3Snž.
X
|
hν, Et3 N |
+ |
|
|
i-PrOH |
||
|
|
||
|
O |
O |
|
O |
|
||
(53) |
(54) |
||
X = OTs, OMs, O2 CCF3 , Br |
|||
>95% |
(33) |
||
|
|
||
X |
|
|
•O |
− |
•O |
O |
• |
|
|
|
|
|
|
|
|
|
(55) |
|
|
|
54 |
|
|
|
|
|
• |
|
|
|
53 |
|
|
|
|
O |
|
|
|
|
|
(56) |
SCHEME 35
Little’s group has reported electroreductive cyclizations involving ˛,ˇ-unsaturated esters (equation 34)96. Based upon CV measurements, the conjugated ester rather than the aldehyde or ketone is suggested to be the electron acceptor. Although direct cyclization of

1316 |
Daniel J. Berger and James M. Tanko |
radical anion 58 was suggested initially, a more detailed mechanistic study suggests that cyclization actually occurs via anion 59 (Scheme 36)97.
CHO
|
|
(57) |
|
|
O |
57 |
e− |
H |
|
|
•
+
‘H ’
O
H
•
e−
 O
O
H
−
(59)
CO2 Me
e−
CH3 CN
Et4 NOTs
+
‘H ’
O−
OCH3
O
OCH3
O
OCH3
SCHEME 36
OH
CH2 CO2 Me
(34)
trans:cis (1.8:1)
 O
O
H O•
−OCH3
(58) |
|
|
|
|
|
|
|
|
−O H |
O• |
|
|
|
OCH3 |
− |
H |
O |
O |
|
OCH3
The preparation of three-, fiveand six-membered rings in modest yield by the electrochemically induced coupling of activated olefins with dihalides has been reported (equation 35)98. Use of an aluminum anode was critical for the formation of cyclized product. Application of this technique to the preparation of four-membered rings was unsuccessful.
MeO2 C |
CO2 Me + |
X-(CH2 )n-X |
|
MeO2 C |
CO2 Me |
e − |
(35) |
||||
|
|
n = 1, 3, 4 |
|
|
(CH2 )n |
|
|
|
|
|
|

22. Radical anions and cations derived from CDC, CDO or CDN groups 1317
(ii) ‘Radical’ reactions of >CDC CDOž . In a series of papers, Mariano reported numerous examples of photochemically-induced interand intramolecular coupling reactions of enones with amines99. These reactions proceed via PIET (Scheme 37) to generate the radical anion/radical cation pair 60. Loss of ‘ZC ’ from the amminium radical cation followed by radical anion/radical coupling leads to the final adduct. An excellent review of this chemistry has recently appeared100.
O
Z
+R2 NCHR′ Z = H, R3 Si
R R
hν
O−
+
/ R2 NCHR′
Z
R R
(60)
−Z+
O−
R R
O
hν
R′
|
CH |
R R |
NR2 |
|
O−
R′
|
CH |
R R |
NR2 |
|
/ R2 NCHR′
SCHEME 37
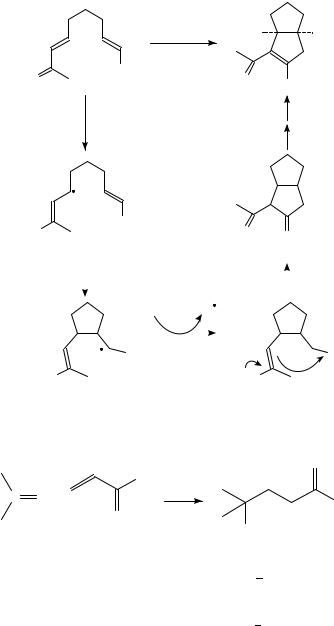
Enholm has reported tandem cyclizations induced by R3SnH (Scheme 38)101. The initial cyclization is a 5-hexenyl-type cyclization of the initially formed radical anion (61 ! 62). After hydrogen transfer from R3SnH, the resulting tin enolate 63 undergoes nucleophilic addition to the nitrile.
(iii) Miscellaneous ketone (aldehyde)/activated olefin couplings. SmI2-promoted coupling of aldehydes or ketones with ˛,ˇ-unsaturated ketones, esters and nitriles have been reported (equation 36). These reactions have been suggested to involve the addition of >CDOž to the activated alkene102, although the inverse (>CDC CDXž C >CDO
!) seems more likely since activated alkenes are more easily reduced than simple aldehydes and ketones. This coupling reaction can also be promoted by Mg in CH3OH103, although mechanistically this is more complicated because the sequence of proton vs
1318 |
Daniel J. Berger and James M. Tanko |
|
||
|
|
n-Bu3 SnH |
H |
H |
|
|
|
|
|
|
|
PhH |
|
|
|
CN |
initiator |
|
|
O |
|
O |
|
|
|
|
NH2 |
||
|
|
|
|
|
R3 Sn
|
CN |
|
|
|
|
|
|
R3 Sn+ −O |
|
O |
|
− |
+ |
||
|
|
|
|
|
N |
R3 Sn |
|
(61) |
|
|
|
|
|
|
|
|
R3 SnH |
R3 Sn |
|
|
|
||
|
|
|
|
||||
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
CN |
|
|
|
|
CN |
|
R3 Sn+ −O |
R3 Sn+ −O |
|
|
|
|||
(62) |
|
|
|
(63) |
|||
SCHEME 38
electron transfers is less clear.
X
Y
C O + |
Sml2 |
|
X |
(36)
Y
OH
IV. RADICAL IONS OF >C=C< CONTAINING COMPOUNDS
A. Chemistry of Radical Cations Derived from Carbon Carbon Double Bonds
As will become evident in this section, in the net transformation from reactant ! product transformations many of the synthetically useful reactions involving >CDC<žC are analogous to those involving neutral, un-ionized carbon carbon double bonds (e.g. the Diels-Alder reaction, oxidation/reduction reactions, nucleophilic addition etc.). However, many of the reactions involving a neutral >CDC< often require the presence of an activating substituent in order to make the alkene more electron-deficient. In a sense, one-electron oxidation of an alkene to its radical cation provides a simple and unique mechanism for increasing the electrophilic (and, of course, radical) properties of
22. Radical anions and cations derived from CDC, CDO or CDN groups 1319
a >CDC< group without having to resort to the incorporation of a substituent. Consequently, radical cation pathways provide a useful alternative in cases where classical synthetic methodologies are not applicable.
1. Methods of generation
Cation radicals may be generated by direct or indirect electrochemical oxidation of the molecule of interest, and many such oxidations are synthetically useful. However, several other methods are also available, which fall into two broad categories: thermally-induced electron transfer (TIET) and photo-induced electron transfer (PIET).
Synthetically important TIET acceptors are dominated by various triarylaminium cation radical salts (Ar3NžC )104. Aminium cation radicals may also be generated electrochemically in situ from the corresponding amines105. High-valent complexes based upon Ce(IV), Fe(III) etc. have also proven useful as one-electron oxidizing agents for radical cation generation106.
Photo-induced electron transfer takes advantage of the fact that the excited state of many compounds (typically possessing electron-deficient -systems) are strong oxidants. Important PIET acceptors include the dicyanobenzenes (DCB), -naphthalenes (DCN) and - anthracenes (DCA), but other more exotic PIET acceptors include 2,4,6-triphenylpyrrilium (TPP)107 and N-alkylacridinium salts. The latter compounds (with long-chain alkyl groups) are particularly useful as PIET acceptors in solvents of medium to low polarity108. TPP salts are typically used as PIET acceptors for reactions such as the cation radical DielsAlder reaction, which requires the use of a minute quantity of catalyst. Use of these compounds is applicable to a wide range of dienes109.
2.Reactions involving >CDC<žC
a. Cation radical ‘pericyclic’ reactions. Over the past decade, several examples of pericyclic reactions induced or catalyzed by the removal of an electron from an olefin have been reported. Three reviews by Bauld cover this group of reactions, especially the socalled ‘hole-catalyzed Diels-Alder’ reactions. The first review is primarily concerned with
the initial work in Bauld’s laboratory110. The second111 and third112 are more comprehensive reviews which cover the field of radical cation ‘pericyclic’ reactions in general, through about 1988 and 1989, respectively. After a brief introduction, this section will focus primarily on more recent developments so as not to duplicate material already covered by the existing reviews.
(i) The ‘hole-catalyzed Diels-Alder’ reaction. Bauld reported that treatment of cyclohexadiene with Ar3NžC led to an unusually facile Diels-Alder reaction (Table 6). It was suggested that radical cations were involved as intermediates in a chain process, and the name ‘hole-catalyzed Diels-Alder’ reaction was coined. The proposed mechanism is summarized in Scheme 39.
Since this initial report, there has been considerable controversy over whether these reactions actually involve radical cations. The alternative possibility is a cationic mech-
anism. (In many cases, R3NžC serves as an indirect source of HC113 . However, this Brønsted-acid-catalyzed pathway can be effectively suppressed by the addition of a hindered base.)
More recently, however, it has been suggested that carbocation intermediates might result from the addition of an amminium ion (through the aromatic ring) to a -bond114. Recently, this controversy seems to have been definitively settled: Most hole-catalyzed Diels-Alder reactions actually do involve cation radicals115, except for the addition of tetracyanoethylene to electron-rich alkenes116.

1320 |
|
|
|
Daniel J. Berger and James M. Tanko |
|||||||
|
|
TABLE 6. Diels-Alder |
reaction of |
cycloheda- |
|||||||
|
|
dienea |
|
|
|
|
|
|
|||
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Conditions |
|
|
Yield |
endo:exo |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
200°C, 20 h |
30% |
|
4:1 |
|
|||||
5 |
|
10 mol% Ar3NžC , |
70% |
|
5:1 |
|
|||||
|
|
||||||||||
|
|
0 °C, 15 min |
|
|
|
|
|
|
|||
|
|
a Reference 110. |
|
|
|
|
|
|
|||
|
|
|
|
dienophile + diene |
|
|
|
adduct |
|||
|
|
|
|
|
|
|
|||||
|
adduct + dienophile |
|
|
|
adduct |
+ dienophile |
|||||
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
diene + dienophile  adduct
adduct
SCHEME 39
Quotation marks are used to describe the ‘hole-catalyzed Diels Alder’ reaction because of the fact that these are Diels Alder reactions only in the sense that they yield the same products. Mechanistically, however, they are distinct and are generally considered to occur in a stepwise, rather than concerted, manner. Results obtained from CIDNP experiments suggest that an intermediate, ring-opened radical cation may be involved in the reaction (Scheme 40)117.
+
+ |
+ |
+ |
SCHEME 40
Additional evidence for a stepwise pathway is provided by the fact that a ‘twostep’ Diels Alder reaction is observed, in which a formal [2 C 2] reaction gives a vinylcyclobutane (64) which then rearranges to the formal [4 C 2] product (Scheme 41, An D p-CH3OC6H4)118. It has been shown that orbital symmetry control does not operate in these reactions: Symmetry-allowed and symmetry-forbidden reactions may take place with equal facility depending upon the conditions119. It has also been shown that the obtention of formally [4 C 2] or [2 C 2] products depends on many factors, including solvent and whether it is the diene or the dienophile which is ionized120.
Typically, electrochemical oxidation of cyclohexadiene gives only polymer121. However, when extreme care is used a low yield (38%) of the Diels Alder adduct with p-methoxystyrene is obtained, with an enhanced endo/exo ratio compared to the same reaction using PIET (Scheme 42)122. For most purposes PIET gives sufficiently high