

Supplement A3: The Chemistry of Double-Bonded Functional Groups. Edited by Saul Patai Copyright 1997 John Wiley & Sons, Ltd.
ISBN: 0-471-95956-1
CHAPTER 15 |
|
|
Hydrogenation of compounds |
|
|
containing C=C, C=O |
|
|
and C=N bonds |
|
|
MARTIN WILLS |
|
|
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK |
|
|
Fax: C44 C 1203 C 524112; e-mail: m.wills@warwick.ac.uk |
|
|
I. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
782 |
|
II. HYDROGENATION OF CDC BONDS . . . . . . . . . . . . . . . . . . . . . . . |
782 |
|
A. Homogeneous Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
782 |
|
1. |
General comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
782 |
2. |
Diastereoselective reductions . . . . . . . . . . . . . . . . . . . . . . . . . |
786 |
3. |
Enantioselective reductions . . . . . . . . . . . . . . . . . . . . . . . . . . . |
789 |
B. Heterogeneous Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
805 |
|
1. |
General comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
805 |
2. |
Diastereoselective reductions . . . . . . . . . . . . . . . . . . . . . . . . . |
806 |
3. |
Enantioselective reductions . . . . . . . . . . . . . . . . . . . . . . . . . . . |
809 |
III. HYDROGENATION OF CDO BONDS . . . . . . . . . . . . . . . . . . . . . . |
810 |
|
A. Homogeneous Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
810 |
|
1. |
General comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
810 |
2. |
Diastereoselective and enantioselective reductions . . . . . . . . . . . . |
811 |
B. Heterogeneous Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
822 |
|
1. |
General comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
822 |
2. |
Diastereoselective reductions . . . . . . . . . . . . . . . . . . . . . . . . . |
822 |
3. |
Enantioselective reductions . . . . . . . . . . . . . . . . . . . . . . . . . . . |
822 |
IV. HYDROGENATION OF CDN BONDS . . . . . . . . . . . . . . . . . . . . . . |
825 |
|
A. Homogeneous Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
825 |
|
1. |
General comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
825 |
2. |
Diastereoselective and enantioselective reductions . . . . . . . . . . . . |
825 |
B. Heterogeneous Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
828 |
|
1. |
General comments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
828 |
2. |
Diastereoselective and enantioselective reductions . . . . . . . . . . . . |
828 |
V. CONCLUSION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
830 |
|
VI. REFERENCES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
831 |
|
781

782 |
Martin Wills |
I. INTRODUCTION
This chapter will review developments in the area of hydrogenation of CDC, CDO and CDN bonds which have been reported in the last ten years or so, thereby providing an update of past volumes in this series. In the area of asymmetric hydrogenation, one outstanding publication provides a comprehensive survey of the area up to 19851. The emphasis will be on synthetic implications rather than the detailed mechanistic analysis of the reactions under study. However, important developments in our understanding of the mechanism of hydrogenation will be described. The synthesis of hydrogenation ligands will not be described in detail, unless the method discussed is a very general one. Coverage will be restricted to the addition of hydrogen across double bonds rather than reduction by other methods such as hydride reagents. Transfer hydrogenation will be included in the review.
Many of the major developments in hydrogenation chemistry in the last decade have been in the area of asymmetric catalysis, and the level of coverage in this review will reflect this important area. In preparing this review it became apparent that many specific catalytic systems may be employed for the reductions of several different classes of double-bonded substrate. To afford maximum utility to the reader as a reference text, the contents have been arranged by type of double bond reduced.
Attention is drawn at this point to a number of excellent reviews of hydrogenation reactions which have recently been published and which will be cited throughout this text.
II.HYDROGENATION OF C=C BONDS
A.Homogeneous Methods
1. General comments
The |
|
high level of research in methods for homogeneous hydrogenation of |
carbon |
|
carbon double bonds originates mainly from the use of the chlorotris(triphenyl- |
|
phosphine) rhodium(I) complex which was first reported by Wilkinson2 and others3,4 in 1965. This review will not seek to define the reaction profile of Wilkinson’s catalyst, since this has been done adequately elsewhere5,6. The Wilkinson catalyst has a very wide substrate range, although less hindered alkenes are reduced more rapidly. It has been demonstrated that the reduction of an alkene takes place in all cases with a syn selectivity, i.e. hydrogen is added to one face of the double bond.5 7 Many modified variants of the Wilkinson catalyst have been reported, some of which benefit from improved substrate selectivity8 18. The chloride atom may be replaced, for example with another halide8, or hydride12b, as may the phosphines. The latter modification provides the greatest scope for productive catalyst modification; dimeric or trimeric phosphines may be employed, including homochiral ligands, as will be described in more detail in a later section. Trialkylarsine and -stibine ligands11 may also be employed in place of phosphines to give active hydrogenation catalysts (Figure 1).
Aqueous solubility may be imparted on the complex most commonly by the incorporation of sulphonic acid groups into the aromatic rings of the phosphine14,19. This method benefits from the particular advantage that it does not interfere with the desirable structural features of many valuable ligands, particularly chiral ligands, as will be described in later sections. Whilst sulphonic acids provide anionic groups to aid solvation, cationic groups based on quaternary amine20 or phosphine21 salts may be employed to the same effect. Another class of water-soluble hydrogenation complexes to be reported recently are those derived from the 1,3,5-triaza-7-phosphaadamantane ligand 122. Complexes of this ligand with rhodium or ruthenium have been shown to be highly effective water-soluble ligands for the hydrogenation of CDC and CDO bonds and for the reduction of enones to

15. Hydrogenation of compounds containing CDC, CDO and CDN bonds 783
Rh
2 |
|
4 |
|
8a |
8a |
|
|
|
|
||||
[RhCl(PPh3)3] |
[RhBr(PPh3)3] |
[RhI(PPh3)3] |
|
|||
[Rh(NO)(PPh3)3]9 |
[Rh(OCOMe)(PPh3)3]10 |
[RhCl(AsPh3)3]11 |
[RhCl(SbPh3)3]11 |
|||
|
|
12b |
|
13 |
14c |
|
[Rh(H)(CO)(PPh3)3] |
[RhCl3(py)3] |
[RhCl(TPPTS)3] |
|
|||
[Rh(COD)(MeCN)2]BF415 |
|
|
(TPPTS D P(m-SO3H)C6H4) |
|
||
|
|
|
|
|||
[Rh2( -Cl)2(CO)2CH2(PPh2)2]C 16 |
[RhCl(py)2(DMF)(BH4)]Cl17 |
|
||||
[Rh6(CO)10(PPh3)6]18 |
|
|
|
|
||
Ru |
|
|
|
|
||
[RuCl2(PPh3)3]26,5b |
[RuHCl(PPh3)3]26 |
[RuH2(PPh3)4]27 |
|
|||
[RuH(NO)(PPh3)3]28 |
[RuCl2(AsPh3)3]29 |
[Ru(OAc)2(PPh3)2]30 |
|
|||
[RuCl(Ph2PCH2CH2PPh2)(MeCN)3]PF631 |
[Ru3(NCO)(CO)10] 32 |
|
||||
|
|
34d |
|
|
|
|
Os [OsHBr(CO)(PPh3)3] |
|
|
|
|
||
Ir [IrCl(CO)(PPh3)3]25e
Co[CoH3(PPh3)3]35
Pt [PtCl2(PPh3)2]37
FIGURE 1. Organometallic complexes used in hydrogenation reactions a Higher activity than chloro compound.
b Terminal alkenes are reduced selectively. c Water and air stable complex.
d Suitable for reduction of conjugated dienes to monoenes. e Vaska’s compound.
N
P
 N
N
N
(1)
aldehydes or ketones. In certain cases the use of a two-phase system is advantageous. The attachment of water-soluble sugar units may also be employed as a strategy to aid water solubility23. The area of water-soluble ligands has been extensively reviewed recently24.
Whilst rhodium complexes have perhaps been the most widely studied reagents for homogeneous hydrogenations, complexes of many other metals have been demonstrated to be highly valuable catalysts. From the point of view of synthetic chemistry complexes based on iridium6,25, ruthenium6,26 32 and osmium6,3,34 are of particular importance and will be discussed in detail throughout this report. Whilst a comprehensive summary of available catalysts is not possible in a review of this type, representative examples of hydrogenation complexes are given in Figure 1. Some of the metals, for example palladium and platinum, are also commonly associated with heterogeneous catalysis, and will be discussed in a later section.
Recent years have seen the introduction and development of metallocene complexes based on early transition metals for hydrogenation. Of complexes of the general formula
784 |
Martin Wills |
[Cp2MR1R2] (R1, R2 D H, alkyl) those derived from zirconium, titanium and hafnium have been most exploited6,38. The catalysts are most conveniently prepared by the action of an excess of a Grignard reagent upon zirconium dichloride complexes39. Many applications of this class of reduction have been reported, including the selective reduction of conjugated dienes to alkenes40, and the mechanism has been studied in some detail39,40. Hydrozirconation has been employed as a method for the reduction of C60 to C60H241. The well-defined structure of the cyclopentadienyl groups and their homochiral derivatives have led to applications of metallocene complexes to asymmetric synthesis, as will be discussed in later sections.
Actinide and lanthanoid complexes have been employed for hydrogenation reactions, for which they often generate dramatic rate increases and high numbers of turnovers42. Many of these complexes exhibit good selectivity for preferential reduction of the less hindered alkene in situations where more than one is present in a substrate (Scheme 1)43.
i
Reagents:(i) 1% [Cp*2 , YMe (THF)] H2
SCHEME 1
Reductions of conjugated alkenes to saturated hydrocarbons may be achieved using a number of methods6. The reduction of dienes conjugated to aromatic rings has been achieved using a binuclear palladium complex pretreated with oxygen (Scheme 2)44. Reductions which stop at the alkene stage are in principle more valuable since a functionality is retained in the molecule. Again there have been several reported methods which achieve this including the use of ferrocenyl amino sulphide and selenide complexes45 and by tricarbonyl chromium arene complexes46. Isomerization of the remaining alkene is often observed and optimal conditions can often only be achieved by careful modifications of the conditions. Many of these processes have been described in detail elsewhere6.
Ph |
i |
Ph |
Ph |
43% |
Ph |
Reagents:(i) 1% [(ButPh)Pd(PBut2 )]2 , THF, rt, 1 atm H2 , 15 min
SCHEME 2
In general, electron-poor double bonds are hydrogenated most rapidly in homogeneous catalytic reactions. This observation greatly facilitates the selective reduction of carbon carbon double bonds in enones and related compounds. The catalyst developed by Wilkinson, [RhCl(PPh)3], has a wide substrate scope in this respect2,6,47, although in the case of aldehydes it can also promote decarbonylation reactions48. One of the most impressive examples of selectivity is in the reduction of unsaturated nitro compounds, without reduction of the nitro group itself, which is normally highly susceptible to reduction48a. The rhodium complex [(PCy3)2Rh(H)Cl2] has been employed in a chemoselective biphasic enone reduction system which is highly selective for the CDC bond49. Catalysts based on ruthenium ([RuCl2(PPh3)3]26b,c and osmium [OsHBr(CO)(PPh3)3]34 work well in the reduction of enones to saturated ketones. Crabtree’s catalyst, [Ir(COD)(PCy3)(Py)]PF6
15. Hydrogenation of compounds containing CDC, CDO and CDN bonds 785
and related complexes, are especially effective for this application50 as are certain arene chromium tricarbonyl complexes51. Certain cobalt complexes have been used as catalysts for the reduction of enones to saturated ketones6. In one recent report the use of cyclodextrin as a phase transfer catalyst in conjunction with hydridopentacyanocobaltate proved to be a highly effective combination for the reduction of ˛,ˇ-unsaturated acids to the saturated derivatives52a. Other methods are described in detail elsewhere6.
The selective reduction of ˛,ˇ-unsaturated carbonyl compounds can be achieved using a palladium complex, [(t-Bu2PH)Pd(t-Bu)2]2, pretreated with oxygen53. Vinylic sulphones and phosphates can be reduced selectively to the saturated products using the same remarkable catalyst54 as can double bonds adjacent to epoxides. In the latter case the epoxide remains undamaged55.
Hydrogenation of aromatic rings will not be described in detail in this review since it is mostly concerned with isolated double bonds. This is however an area of active research, as recently demonstrated by the use of catalytic niobium complexes for the selective hydrogenation of aryl phosphines (Scheme 3)56.
i
P P
3 |
3 |
Reagents:(i) [Nb(OC6 H3 Pri2 )2 ], 4 BunLi, ...
SCHEME 3
Transfer hydrogenation provides a useful alternative to the use of high-pressure hydrogen gas; hydrogen is in this case usually provided by an alcohol such as isopropanol or by formic acid57. Many examples will feature throughout this review, however at this stage simple reductions of the CDC bond in enones will be featured (Scheme 4). The use of microwaves can greatly accelerate this process58. Transfer hydrogenation without the use of a transition metal catalyst can also be achieved using 9,10-dihydroanthracene as a source of the hydrogen59. In this case it is likely that a radical mechanism operates. Transfer hydrogenation of Buckminsterfullerene (C60) and its derivatives has been achieved using this method60. Although a mixture of products is formed, it is possible to maximize the formation of certain reduction species, such as C60H36.
|
O |
|
O |
|
|
i |
|
Ph |
Ph |
Ph |
Ph |
Reagents:(i) (RuCl2 (PPh3 )3 ], PhCH2 OH, 180 °C, 1 h
SCHEME 4
The reactivity of a transition metal catalyst can be modified by redox modification of its oxidation level. In a remarkable example the complex 2, which is formed by the hydrogenation of a precursor complex, may be switched between the C1 and C2 from by the use of an electrode. The C2 form is highly effective at the hydrogenation of alkenes, whilst the C1 complex is more effective for other applications such as the hydrosilylation of ketones52b.
786 |
Martin Wills |
|
|
Ph2 |
2+/+ |
|
P |
|
|
Co |
RhS |
|
|
|
|
P |
|
|
Ph2 |
|
|
(2) |
|
2. Diastereoselective reductions
Many functional groups can co-ordinate to the metal of a hydrogenation catalyst, thereby playing an important part in the directing of diastereoselective reductions of proximal double bonds61. Particularly effective groups in this respect are alcohols, amides, ester and carboxylic acids. Many of these processes have been studied in great detail.
Looking first at alcohol-directed reductions62, it is apparent that there have been many studies of the reduction of allylic and homoallylic alcohols using both the neutral and cationic reduction complexes based on rhodium, iridium etc. In the case of cyclic substrates where an alcohol is located on one side of a ring, the hydrogen is simply delivered cis to the alcohol function63. This is illustrated by key reduction steps in the synthesis of monensin (Scheme 5)64 and the marine natural product arenarol (Scheme 6)65. In each
O |
O |
|
O |
O |
|
i |
|
|
|
BnO |
OH 96% |
BnO |
|
OH |
|
O |
|
|
O |
|
H |
|
|
H |
Reagents:(i) [Rh(COD)Diphos] BF4 , 640 psiH2 |
|
|
|
|
|
SCHEME 5 |
|
|
|
O O O O
i
H
OH |
OH |
|
|
Reagents:(i) [Ir(COD)(PCy3 )py]BF4 , 1000 psiH2 |
|
SCHEME 6 |
|
15. Hydrogenation of compounds containing CDC, CDO and CDN bonds 787
case a cationic complex, based on rhodium and iridium respectively, was employed. Although neutral compounds often work very well in this type of process, the use of the correct cationic complex can be critical in some cases for the full control of diastereoselectivity66.
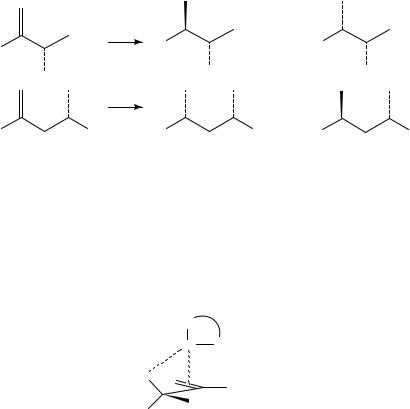
For the reduction of acyclic allylic and homoallylic alcohols a high level of diastereocontrol can be generated in a predicted direction. The general directing effects observed are summarized in Scheme 7 using as an example a cationic rhodium complex as catalyst67. In the case where there is an ˛-chiral centre bearing a co-ordinating group the selectivity is generally observed to be anti, as illustrated. The product formation is believed to take place via a structure such as that illustrated in Figure 2, in which steric eclipsing effects etc are minimized62,68. Although asymmetric reactions will be described in depth in the next section, the potential for kinetic resolution of racemic substrates by matching the effects of chiral ligands to the stereodirecting effects of substituents will be highlighted here. In the example of the carbamate reduction shown in Scheme 8 (the stereodirecting effect is illustrated using an achiral ligand), enantioselectivity ratios (i.e. relative rate of reaction of each enantiomer) of around 20:1 were typical using the chiral ligand DiPAMP (see below) with rhodium in an equivalent cationic complex62,68.
|
|
i |
|
|
|
Ph |
|
Ph |
+ |
Ph |
|
|
|
|
|
|
|
|
|
HO |
97 :3 |
HO |
|
|
HO |
|
|
||
|
|
|
|
|
|
|
|
ii |
+ |
|
|
|
|
|
|
|
|
Ph |
OH |
Ph |
OH 88 :12 |
Ph |
OH |
Reagents:(i) [Rh(NBD)(Diphos)]BF4 , H2 , CH2 Cl2 , (ii) [Rh(NBD) (Diphos)]BF4 , H2 , THF |
|
||||
|
|
SCHEME 7 |
|
|
|
The complexes may be applied to some very demanding and complex transformations as illustrated by a key step in the synthesis of Bafilomycin A1 (Scheme 9), in which a neutral rhodium catalyst was employed69. In contrast, a cationic iridium-based complex was the catalyst of choice in an exacting selective homoallylic alcohol reduction to achieve a vital inversion of configuration in the synthesis of Brevetoxin B70. Functionalized alkenes may also be effectively reduced, significantly trialkylstannanes which
P
Rh P
O
Ph,CO2 Me
R
FIGURE 2. Directing effects in diastereoselective reductions
788 |
Martin Wills |
MeO2 C |
|
|
|
|
|
i |
MeO2 C |
|
||
|
HN |
|
|
|
|
|
|
|
HN |
|
|
O |
|
|
|
|
|
|
|
|
O |
Reagents:(i) H |
, [RhPh PCH |
CH |
PPh ]+ |
|
|
|
||||
2 |
2 |
2 |
|
2 |
|
2 |
|
|
|
|
|
|
|
|
SCHEME 8 |
|
|
||||
|
|
|
|
|
|
|
|
|
OMe |
|
|
|
|
|
|
|
|
|
|
|
OBn |
O |
O |
|
|
O |
|
|
OH |
OTBS |
|
|
|
|
|
|
i |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OMe |
|
|
|
|
|
|
|
|
|
|
|
OBn |
O |
O |
|
|
O |
|
|
OH |
OTBS |
|
|
|
|
|
|
>99:1 d.s. |
|
|
|
|||
Reagents: (i) [RhCl(PPh3 )3 ], 15 bar H2 ,16 h |
|
|||||||||
|
|
|
|
SCHEME 9 |
|
|
||||
OH |
Bu |
|
|
|
|
|
|
|
OH |
Bu |
C6 H11 |
SnBu3 |
|
|
i |
C6 H11 |
|
SnBu3 |
|||
|
|
|
|
|
||||||
Reagents (i) [Rh(NBD) (PPh2 CH2 CH2 PPh2 )]BF4 , H2
SCHEME 10
furnish synthetically valuable and highly diastereoisomerically enriched (60:1 to >500:1) stannanes (Scheme 10)71. Analogous transformations of trialkylsilyl substituted double bonds may also be achieved71b. Although the directing effect of an allylic alcohol is poor in this sense, a series of careful experiments have served to demonstrate that the sulphur chiral centre of vinylic sulphoxides can override its effect72.
15. Hydrogenation of compounds containing CDC, CDO and CDN bonds 789
Amines can direct the reductions of proximal alkenes, as illustrated by a recent example of an enamine reduction to a diamine using the Crabtree iridium catalyst73. Esters and carboxylic acids, although less effective than alcohols, also direct stereoselective reduction reactions (Scheme 11)6,74. As in previous examples cationic rhodium and iridium complexes are favoured. Amides are also good directing groups, and actually slightly superior to esters due to better electron-donating qualities75. Directing effects of co-ordinating groups proximal to double bonds is critical to the success of asymmetric hydrogenations and several examples will feature in the next section.
CO2 Me |
CO2 Me |
CO2 Me |
|
i |
i |
Reagents: (i) [Rh(NBD)(PPh2 CH2 CH2 PPh2 )]BF4 , H2 or [Ir(COD)(PCy3 )py]PF6 , 1 atm H2
SCHEME 11
3. Enantioselective reductions
Recent years have witnessed an explosion in research into asymmetric homogeneous hydrogenations, and this will be reflected in the coverage featured in this section of this review. Several other excellent reviews have been published in this area1,6,76. Horner and Knowles were the first to report that the use of chiral phosphine complexes with rhodium(I) was effective at asymmetric hydrogenation reactions77,78. Later, several researchers found that chelating diphosphines were more convenient ligands and two of the earliest of these, DIPAMP (P1)79 and DIOP (P2)80, have proved to be as good in many respects as ligands reported at later dates. Each is designed with a different principal in mind; in the first the proximity of a chiral centre as close to the metal as possible and in the second the use of a chiral ‘array’ of phenyl groups to create a chiral environment. Many ligands have employed the same design characteristics since these were reported. Whilst it is impossible in a review of this type to be able to list all of the chiral phosphines which have been reported, a range of representative examples is given in Figure 3. Encyclopeadic surveys are available from other sources1,76. In addition to illustrating the great diversity of available ligands, the selection includes some very recently reported phosphines. Many of these will be referred to in the following discussion and will feature in future sections of this review.
The methods for the preparation of these ligands will not be discussed in detail in this review, however the introduction of the phosphine unit is most commonly achieved using a nucleophilic source of phosphine, e.g. Ph2PLi etc. The synthesis of C2 symmetric ligands such as DiPAMP requires a key coupling reaction of two monophosphine precursors. As for most phosphines, oxidation by oxygen is a potential hazard. Whilst this can be avoided by careful handling, temporary protection of the phosphine as a borane complex has been very commonly employed during the synthesis of many of the chiral ligands
shown below104,105. |
|
Although asymmetric hydrogenation |
has found many applications in synthesis, |
the most intensely investigated area is |
almost certainly the hydrogenation of ˛-N- |
acylaminoacrylates (Scheme 12)1,6,76,106. In most cases modest pressures (1 atm) of hydrogen are required, and low quantities (<1 mol%) of catalyst, which may frequently be conveniently formed in situ by the combination of a ligand with a non-chiral source of rhodium. Whilst rhodium is most commonly the metal of choice for catalysts, BINAP
790 |
|
|
|
Martin Wills |
|
OMe |
|
|
|
|
|
|
|
|
|
H |
PPh2 |
.. |
|
Ph |
|
O |
|
|
|
|
PPh2 |
|
|
P |
P |
OMe |
PPh2 |
|
|
|
|
|
|
||
Ph |
|
|
|
|
|
|
|
|
O |
PPh2 |
|
|
|
|
|
H |
|
|
|
|
|
|
|
(P1) DiPAMP |
79 |
|
(P2) RR DIOP8 0 |
(P3) SS Chiraphos8 1 |
|
|
|
|
|
||
PPh2 |
|
|
|
|
PPh2 |
PPh2 |
|
|
|
|
|||
|
|
|
|
|
|
|
PPh2 |
|
|
|
|
PPh2 |
PPh2 |
|
|
|
|
|
||
|
|
|
|
|
||
8 2 |
|
(P5) R-BINAP8 3 |
(P6) RR Norphos8 4 |
|||
|
||||||
(P4) R Prophos |
|
|
|
|
|
|
Ph2 P |
|
Ph2 P |
|
|
PPh2 |
PPh2 |
PPh2 |
|
|
|
|
|
PPh2 |
|
|
|
|
|
|
|
N |
|
|
N |
|
||
CO2 But |
|
|
CO2 But |
|
||
(P7) BPPM8 5 |
|
(P8) SS PYRPHOS8 6 |
(P9) Skewphos8 7 |
|||
Ph |
|
|
|
|
PCy2 |
|
|
|
|
|
|
PPh2 |
|
O |
|
|
|
|
|
|
O |
|
|
|
|
PPh2 |
|
O |
|
OPh |
|
|
|
|
|
|
|
|
|
||
Ph2 PO |
|
|
|
|
|
|
|
|
|
|
|
PPh2 |
|
OPPh2 |
|
|
Fe |
|||
|
|
|
||||
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
Cp |
|
|
(P10)8 8 |
|
|
|
(P11) R-Josiphos8 9 |
(P12)9 0 |
|
FIGURE 3. Representative chiral phosphine ligands