

Orbital Interaction Theory of Organic Chemistry
.pdfSTEREOTOPIC RELATIONSHIPS OF GROUPS IN MOLECULES |
9 |
Figure 1.4. Flow chart for deciding stereotopic relationships between pairs of groups.
STEREOTOPIC RELATIONSHIPS OF GROUPS IN MOLECULES
Many of the ideas espoused in this and the next section are due to the work of Mislow [29]. For an alternative discussion of the concepts introduced in this section, see reference 30. The reader is also directed to excellent texts by Juaristi [31] and by Eliel and Wilen [32].
The concepts used to describe relationships between pairs of molecules may readily be extended also to pairs of groups within a molecule [17]. This is particularly useful in determining the appearance of an NMR spectrum or the possibility of selective reaction at similar functional groups. Regions (such as faces of planar portions) around molecules may be similarly classi®ed. The same relationships could also be applied to (groups of ) atomic orbitals within the molecule. These are collectively referred to as ``groups'' for the purpose of the ¯ow chart in Figure 1.4. From the analysis of Figure 1.3, three broad groupings of properties emerge:
1.Homotopic Groups Not distinguishable under any conditions, chiral or achiral. To have homotopic groups, a molecule must have a ®nite axis of rotation. Thus the only molecules which cannot have homotopic groups are those whose point groups are C1; Cs; Ci, and Cyv.
2.Enantiotopic Groups The same in all scalar properties, distinguishable only under chiral conditions.
3. Constitutionally Heterotopic and Diastereotopic Groups Di¨er in all scalar properties and are distinguishable under any conditions, chiral or achiral. Asymmetric molecules cannot contain homotopic or enantiotopic groups, only diastereotopic or constitutionally heterotopic groups.

10 SYMMETRY AND STEREOCHEMISTRY
Groups may be compared by internal comparison (groups in the same molecule) or by external comparison (groups in di¨erent molecules).
One can also compare faces of a molecule in the same way as groups, since the comparison actually applies to environments. Thus, the two faces of the carbonyl groups of aldehydes, unsymmetrical ketones, esters, and other acid derivatives are enantiotopic. Reaction at the two faces by a chiral nucleophile will take place at di¨erent rates, resulting in asymmetric induction.
Exercise 1.3. Verify the following group designations:
Homotopic groupsÐ…H1; H4†, …H2; H3†, …H5; H6†
Enantiotopic groupsÐ…H1; H2†, …H3; H4†, …H1; H3†, …H2; H4†
Constitutionally heterotopic groupsÐany of H1; . . . ; H4 with
H5 or H6
F1 and F2 are homotopic faces.
There are no diastereotopic groups in this molecule.
Exercise 1.4. Verify the classi®cation of the pairs of groups in tricyclo[3:1:0:02; 4]hexane.
HomotopicÐ…H1; H6†, …H2; H5†, …H3; H7†, …H4; H8†
EnantiotopicÐ…H3; H4†, …H3; H8†, …H4; H7†, …H7; H8†
DiastereotopicÐ…H1; H2†, …H1; H5†, …H2; H6†, …H5; H6†
Constitutionally heterotopicÐ…H1; H3†, …H1; H4†, …H2; H8†; . . .
Exercise 1.5. Compare all of the groups and faces of the trans-3,4-dimethylcyclopenta- nones below, by both internal comparison and external comparison.
ASYMMETRIC SYNTHESIS AND STEREOCHEMISTRY
Asymmetric synthesis is any synthesis that produces enantiomerically or diastereomerically enriched products. This is the expected result if enantiomerically enriched chiral substrates are employed. Of interest here are asymmetric syntheses where the reactants are either achiral or chiral but racemic. Many examples of this type are collected in volumes edited by Morrison [33]. The ®rst example of an asymmetric synthesis involved use of the chiral, optically pure base brucine in a stereoselective decarboxylation of a diacid with enantiotopic carboxyl groups [34]:
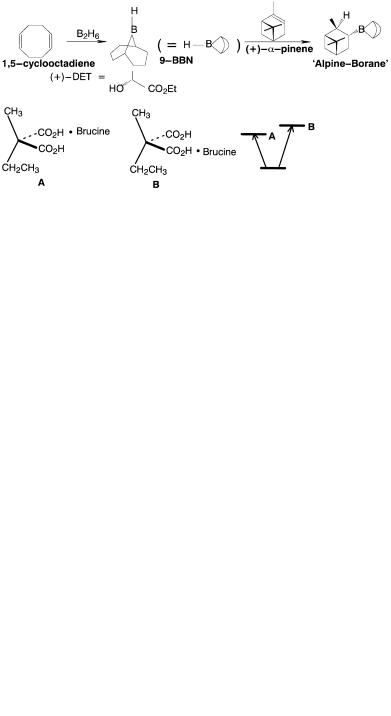
ASYMMETRIC SYNTHESIS AND STEREOCHEMISTRY |
11 |
The monobrucine salts A and B are diastereomeric and therefore di¨erent in all properties, including activation energy for decarboxylation. A carbon atom which contains two enantiotopic groups is prochiral. E½cient stereoselection or asymmetric induction requires tight binding of the chiral reagent to the achiral substrate. In addition, there should be a large steric or stereoelectronic distinction between the groups in both substrate and the chiral reagent. For this purpose, the distinction between methyl and ethyl groups in Markwald's experiment shown above is less than ideal. The tight binding requirement can be satis®ed by the use of transition metals to which chiral auxiliaries are attached as ligands. One example, the Katsuki±Sharpless epoxidation of allylic alcohols [35], serves to illustrate the principles:
Both the allylic alcohol and tert-butyl hydroperoxide are achiral, but the product epoxide is formed in high optical purity. This is possible because the catalyst, titanium tetraisopropoxide, forms a chiral (possibly dimeric [36]) complex with resolved diethyl tartrate […‡†-DET] which binds the two achiral reagents together in the reactive complex. The two enantiotopic faces of the allylic double bond become diastereotopic in the chiral complex and react at di¨erent rates with the tert-butyl hydroperoxide. Many other examples may be found in recent reviews [31, 37±39].
The ®eld of organoboron chemistry pioneered by Brown [40] also provides a wealth of excellent transformations. Consider the asymmetric reduction of carbonyl compounds by Alpine-Borane [41]. Alpine-Borane is prepared by the following sequence:

12 SYMMETRY AND STEREOCHEMISTRY
In the second step, achiral 9-borabicyclo[3.3.1]nonane (9-BBN) adds to the less hindered diastereotopic face of a-pinene to yield the chiral reducing agent Alpine-Borane. Aldehydes are rapidly reduced to alcohols. The reaction with deuterio-Alpine-Borane, which yields (R)-a-d-benzyl alcohol in 98% enantiomeric excess (ee) reveals a very high degree of selectivity of the enantiotopic faces of the aldehyde group in a crowded transition state:
As a consequence of steric congestion in the transition state, ketones generally require high pressures to increase the reaction rate but yield optically active secondary alcohols in high ee. Thus, acetophenone yields 100% ee. of (S)-1-phenylethanol at 2000 atm:
Many instances of stereospeci®c selection of enantiotopic groups or faces may be found in nature. One such is extracted from the tricarboxylic acid cycle and is shown in Exercise 1.6. At each step, achiral reactants are transformed to achiral products with high stereospeci®city!
Exercise 1.6. Analyze the following sequence from the tricarboxylic acid cycle (*C denotes isotopically labeled carbon):
NMR AND STEREOCHEMISTRY
Nuclear magnetic resonance chemical shift di¨erences can serve as an indicator of molecular symmetry. If two groups have the same chemical shift, they are isochronous. Isochrony is a property of homotopic groups and of enantiotopic groups under achiral conditions. Diastereotopic or constitutionally heterotopic groups will have di¨erent chemical shifts (be anisochronous), except by accidental equivalence and/or lack of su½cient resolution.

NMR AND STEREOCHEMISTRY |
13 |
To be anisochronous, (1) groups may not be related by symmetry, taking into consideration internal motions which are rapid on the NMR time scale, and (2) there must be su½cient ®eld gradient so that the di¨erence is observable.
For homotopic groups, chemical shifts are indistinguishable in chiral or achiral solvents, that is, the groups are isochronous.
Enantiotopic groups are isochronous in achiral solvents and distinguishable (anisochronous) in chiral solvents.
In principle, the enantiotopic protons of bromochloromethane will be anisochronous in a chiral solvent. However, it requires a fair degree of association to make the chemical shift di¨erence visible. This requirement may be satis®ed in hydrogen-bonding solvents:
Thus the enantiotopic methyl groups of dimethylsulfoxide form an A3B3 spin system in 1-phenyl-2,2,2-tri¯uoroethanol; Figure 1.5 [42]:
The hydrogen-bonding association of amino acid esters with 1-phenyl-2,2,2-tri¯uoro- ethanol is su½cient to permit NMR to be used as a method for determining the optical purities of a-amino acids [43].
The same principle is involved in the use of chiral lanthanide chemical shift reagents for the determination of enantiotopic purity [44].
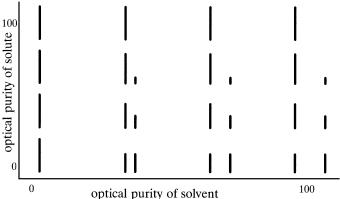
Figure 1.6 illustrates the expected observations when a chiral solute is dissolved in a chiral solvent and optical purities of both vary from zero (racemic) to 100%. When the optical purity of the solvent is increased, the separation of the enantiomer (actually diastereomer in the chiral solvent) signals increases. When the signal separation is su½cient, the optical purity of the solute may be determined by integration of the paired signals. When the solute optical purity is 100%, only a single signal is observed at all solvent optical purities. Recognition of the solute optical purity in the absence of the second
Figure 1.5. Simulated NMR spectrum of methyl alanine in a chiral solvent.
14 SYMMETRY AND STEREOCHEMISTRY
Figure 1.6. E¨ect of solvent and solute optical purity on the appearance of NMR signals of enantiomers or enantiotopic groups (bottom row).
signal requires prior knowledge of the expected pure enantiomer chemical shift under the conditions of the experiment.
Enantiomeric purity is often determined by derivatization with an optically pure chiral agent. For alcohols and amines, a-methoxy-a-tri¯uoromethylphenylacetic acid (MTPA) and a-cyano-a-¯uorophenylacetic acid (CFPA) [45] work well.
SYMMETRY AND STRUCTURAL PARAMETERS
Structural parameters (bond lengths, bond angles, dihedral angles) must be the same in a molecule when they are interconvertible by a symmetry operation, that is, congruent. Conversely, structural parameters cannot be the same in a molecule when they are not congruent. If the structural parameters are not congruent, it is not possible to use symmetry arguments to predict the magnitude of the di¨erence.
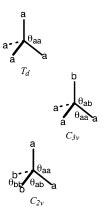
Some relationships between the bond lengths and angles of nominally tetrahedral molecules are shown below. The notation a and b denote groups which are di¨erent in some way. The point groups shown denote the molecular point group. For the relationships to hold exactly, the structures of a and b must be such as to preserve the overall symmetry. The relationships may be approximately obeyed if the denoted point groups are a fair representation of the local symmetry. For example, the ®rst structure will have exactly Td symmetry if a is H, Cl, or Me but not if a is Et, since the ethyl group does not have a threefold axis of symmetry. Equation (1.1) de®nes the tetrahedral angle. Equation (1.2) may be handy for relating the internal bond angle of a threefold symmetric species, such as ammonia, to the out-of-plane angle of the bonds. Equation (1.3) applies to molecules like methylene chloride (CH2Cl2) or cyclopropane.
NOTE ON HYBRIDIZATION |
15 |
cos yaa ˆ ÿ 13 …1:1†
3 sin2 yab ˆ 2…1 ÿ cos yaa† |
…1:2† |
cos yab ˆ ÿcos…21 yaa† cos…21 ybb† |
…1:3† |
Exercise 1.7. What point groups are available for di¨erent orientations of the ethyl groups in the `Td ' structure with a ˆ Et?
NOTE ON HYBRIDIZATION
The concept of hybridization was introduced to provide a mechanism for achieving directionality in bonding, recognizing implicitly that linear combinations of the 2s and some of the three 2p orbitals point in well-de®ned directions relative to other such combinations. Thus if one takes a 1 : 1 linear combination of the 2s orbitals and one of the 2p orbitals (leaving the other two 2p orbitals alone), one obtains two sp hybrid orbitals which are directed at an angle of 180 to each other. As we shall see later, orbitals mix (or hybridize) so as to provide the best overlap for bonding. Mixing the 2s orbital with two of the three 2p orbitals yields three equivalent sp2 hybrid orbitals which are exactly arranged at 120 relative to each other, yielding the familiar trigonal planar pattern of bonds when each sp2 hybrid orbital forms a sigma bond to a di¨erent but identical atom or group. Likewise, four equivalent sp3 hybrid orbitals directed toward the corners of a tetrahedron with equal interorbital angles of 109:47 are obtained when the 2s and all three 2p orbitals are mixed. The one, two and three refer to the ``weights'' of the 2p orbitals relative to the 2s orbital in the sp, sp2, and sp3 hybrid orbitals, respectively. The angles between two equivalent hybrid orbitals are determined by the weights of the 2p : 2s mixture. Conversely, observation of interbond angles of 180 , 120 , and 109:47 between two equivalent (by symmetry) geminal CÐX bonds implies that the carbon atom is using sp, sp2, and sp3 hybrid orbitals, respectively, to form those bonds. Hybridization can be inferred from the observed angles. Since the observed interbond angles are rarely the idealized values 180 , 120 , and 109:47 , it follows that the orbitals are not the idealized hybrids but rather hybrids where the weight of the 2p orbital relative to the 2s orbital is a positive real number, say l2. In this case, a general hybrid orbital, hi, will thave the composition s ‡ li p, which is equivalent to spli2 hybridization. The weight li2 may range from zero to in®nity (pure s to pure p). Normalization of the hybrid orbitals requires that the following relationships hold:

16 |
SYMMETRY AND STEREOCHEMISTRY |
|
|
|
|||||
|
X |
1 |
ˆ 1 |
1 |
|
ˆ s character of hybrid orbital hi |
…1:4† |
||
|
|
|
|
|
|
||||
|
1 ‡ li2 |
1 ‡ li2 |
|||||||
|
i |
l2 |
|
|
|
l2 |
|
|
|
|
Xi |
i |
ˆ 1; 2; 3 |
|
|
i |
ˆ p character of hybrid orbital hi |
…1:5† |
|
|
1 ‡ li2 |
|
1 ‡ li2 |
||||||
In equations (1.4) and (1.5), the sums run over the number of hybridized orbitals. For any pair of hybrid orbitals, hi and hj , the following relationship exists:
1 ‡ lilj cos yij ˆ 0 |
…1:6† |
where yij is the angle between two hybridized orbitals.
Exercise 1.8. What is the hybridization of the carbon orbitals which form the CÐH and CÐC bonds of cyclopropane (HCH ˆ 114 )? Verify that if the carbon hybrids which are used for the CÐH bonds are exactly sp2, then the two equivalent hybrids for the CÐC bonds must be sp5 and the interorbital angle is 101:5 !
Empirically, C13 ÐH spin±spin coupling constants are proportional to the ``s character'' of the hybrid orbital used in the s bond to H:
JCH …cps†A |
500 |
…1:7† |
1 ‡ li2 |
SYMMETRY AND ORBITALS
Symmetry properties of atomic and molecular orbitals will prove useful in a variety of contexts. We will familiarize ourselves with the characteristics of the basic types of orbitals which will be used throughout the remainder of this book. It is not proper to assign a point group label to orbitals because of the phase characteristics, but rather to the charge distribution which would result upon squaring the orbital. The orbital may then be characterized by designating the label of the irreducible representation according to which it transforms within the context of the local or global molecular point group. These attributes are speci®cally described for atomic s, p, and spn (hybrid) atomic orbitals and for molecular orbitals below.
Atomic Orbitals
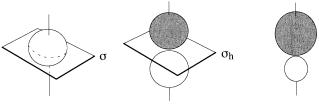
The symmetry characteristics of s, p, and spn (hybrid) atomic orbitals are illustrated in Figure 1.7. Thus the charge distribution due to an electron in an atomic s orbital is spherically symmetric (point group Kh) and the s orbital itself will transform as the totally symmetric irreducible representation. Alternatively, one may assign a label, S or A, which describes the behavior of the orbital under any relevant symmetry operations. For instance, the s orbital does not change sign (phase) upon re¯ection in any plane containing its center or upon rotation through any angle about any axis of symmetry. It is symmetric with respect to any symmetry operation, and this characteristic is conveniently assigned the label S for whichever symmetry operation is considered. On the other hand, the charge distribution due to an electron in an atomic p orbital is dumbbell
SYMMETRY AND ORBITALS |
17 |
…a† |
…b† |
…c† |
Figure 1.7. The symmetry characteristics of (a) s, (b) p, and (c) spn (hybrid) atomic orbitals. The shapes of the electron distributions are similar if one ignores the phases.
shaped (axially symmetric with a horizontal mirror plane, point group Dyh). The p orbital itself will transform as the irreducible representation S‡u ; that is, the p orbital does not change sign (phase) upon re¯ection in any plane containing its principal axis or upon rotation through any angle about the principal axis but does change sign (phase) upon re¯ection across the horizontal mirror plane (its own nodal plane) and rotation about any axis of symmetry (necessarily twofold) contained in that plane. It is symmetric (S) with respect to any of the ®rst set of symmetry operations. It is antisymmetric with respect to any of the second set of symmetry operations, and is assigned the label A for these. Hybrid atomic orbitals spn retain only the axial symmetry of the pure s and p orbitals. The node (boundary separating the two phases of the orbital) is now a curved surface and no longer a symmetry element. The charge distribution belongs to the point group Cyv, and the hybrid orbital transforms as the a1 irreducible representation of
Cyv.
Molecular and Group Orbitals
Let us accept that molecular orbitals (MOs) and group orbitals are both described as linear combinations of atomic orbitals. Exactly how and why this is the case will be seen in Chapter 2 and Appendix A. For the purpose of the present section, proper MOs are those linear combinations which transform as irreducible representations of the molecular point group, that is, are symmetry adapted. Group orbitals are linear combinations which are symmetric or antisymmetric with respect to any local symmetry operations of that part of the molecule which constitutes the group (e.g., a methyl group). At an intermediate level of description, MOs may be thought of as linear combinations of group orbitals. We shall frequently use the term localized orbital. This term has a formal de®nition in the literature of electronic structure theory, but we shall use it in a loose sense to describe a characteristic piece of a true MO or a group MO such as a sigma bond between a particular pair of atoms or an atomic orbital describing a nonbonded pair of electrons. A localized MO may indeed be a proper MO or a group MO which happens to be concentrated in one region of the molecule. More likely, however, a proper MO or a group MO would be described as a linear combination of localized MOs. Some examples of proper MOs and group MOs are shown in Figure 1.8. Notice that the ``proper'' MOs of water which describe the ``lone pairs'' of electrons are inand out-of-phase combinations of the ``rabbit ears'' often pictured in elementary texts. The out-of-phase combination has no s character at all. It is a pure p orbital on the oxygen atom. The same is true of the proper MOs which describe the OÐH bonds.
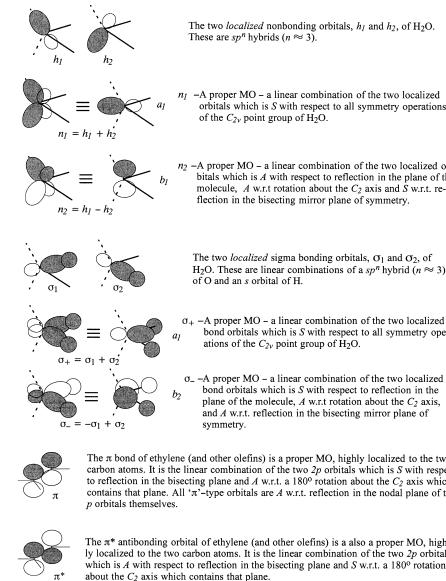
18 SYMMETRY AND STEREOCHEMISTRY
Figure 1.8. Examples of symmetry-adapted (proper) MOs and their constituent atomic or localized orbitals.