

Orbital Interaction Theory of Organic Chemistry
.pdfWHY DOES IT WORK AND WHEN MIGHT IT NOT? |
69 |
which de®nes the p-type orbital is not shown), is indicated by an arrow in Figure 3.23. It is along a line that makes an angle somewhat greater than 90 with the CO bond. Attachment of a Lewis base means addition of electrons to the pCO orbital and is accompanied by loss of the p bond (if both bonding and antibonding orbitals are equally occupied, there is no bond).
WHY DOES IT WORK AND WHEN MIGHT IT NOT?
The application of orbital interaction theory to understand the electronic structures of molecules, as illustrated in the case of the carbonyl group in Figure 3.21 above, is one of two conceptually distinct applications. Characteristics of the bonding and structure of the molecule were deduced from interactions of the group orbitals situated as they would be in the ``®nished'' molecule. This application is in the regime of strong interactions and short distances, the regime in which the foundations of orbital interaction theory, oneelectron theory as exempli®ed by Hartree±Fock theory, have a ®rm footing and a demonstrated record of successes. Since the electrons are con®ned to a small region of space, lack of speci®c account of electron correlation is not serious in that its inclusion would not substantially a¨ect the one-electron description, or the error incurred may be assumed to be reasonably constant when comparing di¨erent conformations.
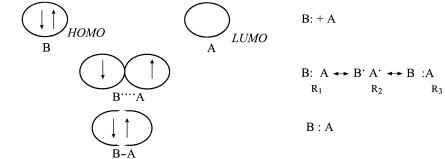
The second broad area of application of orbital interaction theory is in the area of intermolecular interactions, from which many aspects of chemical reactivity may be inferred. This application, as exempli®ed in Figures 3.22 and 3.23, is in the realm of weaker interactions and larger distances, conditions suitable to application of perturbation theories. The dominant orbital interactions in this regime are of the two-electron, two-orbital type, and usually, a single orbital interaction between the HOMO of one molecule and the LUMO of the other is su½cient for the purpose. Unfortunately, the one-electron theoretical foundation for this kind of long-range interaction involving electron transfer is much less sound. Accurate descriptions of weak interactions usually require theoretical procedures which go beyond Hartree±Fock theory for the reason that some account must be taken of the correlation of the electron motions, particularly within the same orbital. The reason for this is easy to understand and is illustrated in Figure 3.24. Prior to interaction (Figure 3.24a), the electrons are localized to the HOMO of the donor, B. At intermediate separations (Figure 3.24b), where the orbitals of A and B overlap, the space available to the electrons is greatly increased and the electrons are able to separate. After bond formation (Figure 3.24c), the space available to the electrons is again restricted. In the intermediate stage (Figure 3.24b), the lack of electron correlation inherent in one-electron theories is most strongly felt. In terms of valence bond structures, MO theory places comparable emphasis on the three structures R1, R2, and R3, whereas, in reality, resonance structure R2 alone most accurately represents the true situation. Resonance structure R2 corresponds to a con®guration in which a single electron has been transferred from B to A. The reactivity of B as a donor (nucleophile) is expected to be correlated with its ionization potential [76]. Likewise, the reactivity of A as a Lewis acid should be correlated with the electron a½nity of A. Post-Hartree±Fock theoretical analyses of heterolytic bond breaking [77] and the reverse, cation±anion combination reactions [78], have elucidated the role of the missing electron correlation. In the valence bond con®guration mixing (VBCM) treatment of Shaik [9, 78], the valence bond con®gurations of reacting species are regarded as ``surfaces'' whose avoided crossing de®nes the barrier to the reaction. The Hartree±Fock model is expected to work well
70 ORBITAL INTERACTION THEORY
…a†
…b†
…c†
Figure 3.24. Stages in the interaction between two molecules, B and A, showing the e¨ects of electron correlation and the equivalent resonance structures: (a) no interaction, the electrons are spin paired and con®ned to the orbital of B; (b) weak interaction (e.g., a transition structure), the electrons can separate into a larger volume of space; (c) strong interaction, a bond is formed and the electron distribution is again con®ned.
when the electronic description involves a single closed-shell con®guration (Figures 3.24a,c) but overestimate the e¨ect of electron±electron repulsion in the extended case (Figure 3.24b). It is not surprising, then, that Hartree±Fock calculations tend to overestimate the energy of the transition state for intermolecular reactions. This failure should not be interpreted as a general failure of the one-electron model as an interpretive device, but rather as an inappropriate energy counting for the MO description due to the fact that electron±electron repulsions are accommodated in an average way. The MO description remains as a powerful and conceptually simple means for understanding the bonding even in the region of the transition state, and such MO descriptions are derivable by the simple rules of orbital interaction theory.
Particulary for the intermolecular case illustrated by Figure 3.24, it is tempting to use experimental studies carried out in the gas phase, or the results of high quality ab initio calculations (carried out in vacuo), as a ``test'' of the validity of orbital interaction theoretical predictions. In the gas phase, one expects to have the ``pure'' orbital interactions, free of the complex contributions from the solvent. This expectation is not justi®ed under some well-de®ned circumstances beyond those described immediately above. The e¨ects of electrostatic interactions, discussed brie¯y above, are unquenched in the absence of an intervening medium. Such interactions, particularly between charged and/or strongly dipolar species, may completely overwhelm the more subtle interactions due to the frontier orbitals. Thus gas-phase experiments and theoretical calculations on ionic or polar molecules may in no way re¯ect on the interactions of orbitals because electrostatic interactions have a very high potential and are very long range. In a solvent of high dielectric constant, these are greatly ameliorated, permitting orbital interactions to dominate. Indeed, it has been shown by ab initio calculations in the presence of a medium (modeled as a continuum) that groups become less electronegative and less hard with increasing dielectric constant [79]. Thus the electronegativity of a methyl group in water is decreased by about 75% relative to its gas-phase value.
Molecular orbital calculations also do not provide an easily interpretable picture in terms of group orbital interactions for several reasons. First, the basis set does not consist of group orbitals but rather AO-like functions from which group orbitals are also constructed. Second, by de®nition, an MO calculation takes into account all possible
WHY DOES IT WORK AND WHEN MIGHT IT NOT? |
71 |
group orbital interactions, and it may be di½cult to identify the dominant two-orbital interaction on which our application of orbital interaction theory depends, a matter of not seeing the forest for the trees. As a case in point, the structures of 1-methylcyclohexyl carbocation as predicted by ab initio MO calculations are readily interpreted in terms of a two-orbital interaction diagram such as Figure 3.9, which describes the hyperconjugative interaction between the cationic site and adjacent CÐH or CÐC sigma bonds. However, evidence of the bonding interaction is completely obscured among the many occupied orbitals of the molecule. One needed to seek evidence of the bonding by examining the antibonding interactions in the more highly localized LUMO [80, 81]!

Orbital Interaction Theory of Organic Chemistry, Second Edition. Arvi Rauk Copyright ( 2001 John Wiley & Sons, Inc.
ISBNs: 0-471-35833-9 (Hardback); 0-471-22041-8 (Electronic)
CHAPTER 4
SIGMA BONDS AND ORBITAL
INTERACTION THEORY
CÐX s BONDS: X FC, N, O, F AND X FF, Cl, Br, I
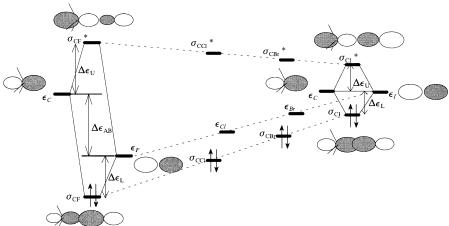
Consider the interaction between two spn hybridized orbitals of unequal energy interacting in a s fashion. The interaction diagram is illustrated in Figure 4.1a. Notice that the hybridization of the spn group orbitals in the s orbital appears to be turned inside out. This is in recognition that much cancellation will occur due to the node in the middle. The lowest energy con®guration has two electrons with energy eL, as shown. Not much can be said about the absolute magnitudes of DeL and DeU, but we are concerned here with trends. The ®rst series (C, N, O, F) involves atoms which are very approximately the same size but di¨ering greatly in electronegativity. The interaction matrix element, hCX, will be essentially constant across the series X ˆ C, O, N, F. However, relatively large changes will occur in the di¨erence in orbital energies, the quantity eC ÿ eX increasing in the series. The consequent energies of the resulting s and s orbitals are shown in Figure 4.2. In each of the two interaction diagrams shown and the two implied in between, the energy of the spn orbital of the C atom is drawn at the same level. The energies of both the bonding s orbital and the antibonding s orbital decrease, being the lowest for the CÐF bond. The greatest polarization also occurs in the case of the CÐF bond. In particular, the low-lying s orbital is highly polarized toward the C end of the bond.
The second series, involving bonding of C to di¨erent halogens (F, Cl, Br, I) involves atoms which are very di¨erent in size and also di¨ering greatly in electronegativity. The halogen ends of the bonds are constructed from 2p, 3p, 4p, and 5p orbitals, respectively. The overlap, and therefore the interaction matrix element, hCX, will decrease very rapidly across the series, X ˆ F, Cl, Br, I. Relatively large changes will also occur in the di¨erence in orbital energies, the quantity eC ÿ eX decreasing in the series. The relative properties and reactivities of CÐX bonds are consistent with dominance of the interaction matrix element (i.e., overlap dependence) in determining the resulting orbital
72

CÐX s BONDS: X FC, N, O, F AND X FF, Cl, Br, I |
73 |
…a† |
…b† |
Figure 4.1. (a) A s bond between atoms of di¨erent electronegativity; (b) homopolar s bond.
energies. The consequent energies of the resulting s and s orbitals are shown in Figure 4.3. The sCX decreases along the series and the sCX bond increases. The predicted trend in sCX bond energies is observed in the bond ionization potentials [82]. The lowest lying s orbital is expected to be that of the CÐI bond.
…a† |
…b† |
…c† |
…d† |
Figure 4.2. The sCX bond and sCX antibond between atoms of the ®rst row: (a) X ˆ C; (b) X ˆ N; (c) X ˆ O; (d ) X ˆ F.
74 SIGMA BONDS AND ORBITAL INTERACTION THEORY
…a† |
…b† |
…c† |
…d† |
Figure 4.3. The sCX bond and sCX antibond between di¨erent halogens: (a) X ˆ F; (b) X ˆ Cl; (c) X ˆ Br; (d ) X ˆ I. No attempt has been made to show the increased size or nodal character of the p orbitals of Cl, Br, or I.
s BONDS: HOMOLYTIC VERSUS HETEROLYTIC CLEAVAGE
Heterolytic Cleavage of s Bonds Involving C or H
In heterolytic bond cleavage, both electrons end up on system B. The energy change associated with the bond dissociation is just the di¨erence between electron energies before …2eL† and after …2eB†, equation (4.1) and Figure 4.1:
|
het |
|
2h |
2 |
|
DE |
ˆ ÿ2…eL ÿ eB† ˆ 2DeL A |
|
AB |
…4:1† |
|
|
DeAB |
||||
The last approximation in equation (4.1) is only really valid for the small-overlap regime, which this is not, but it serves to remind us of the approximately inverse energy dependence of the stabilization. One may ask how DE het would be expected to change in the series CÐC, CÐN, CÐO, and CÐF or the series HÐC, HÐN, HÐO, and HÐF. In the crudest approximation, one may say that the orbitals of C, N, O, and F are all approximately the same size and therefore the interaction matrix element hAB will be approximately the same size for any A±B pair. The dominant factor determining the heterolysis energy therefore is the di¨erence in orbital energies in the denominator, and one has directly the prediction (Figure 4.2) that ease of heterolytic cleavage for CÐX is in the order C > N > O > F. The CÐC bond is least likely to dissociate heterolytically and the CÐF bond the most likely. In an absolute sense, of course, heterolytic cleavage is not a likely process for any of these bonds in the absence of other factors, as discussed below.
Similarly, one may ask how DE het would be expected to change in the series CÐF, CÐCl, CÐBr, and CÐI. The situation is more complicated for this series, since both
s BONDS: HOMOLYTIC VERSUS HETEROLYTIC CLEAVAGE |
75 |
the electronegativity and orbital size vary. Variation in orbital size is approximately proportional to n2, where n is the principal quantum number of the valence shell. The larger the discrepancy in orbital sizes, the smaller is the e¨ective overlap and the smaller is the interaction matrix element, hAB. It should be noted that because of the smaller overlaps, the last approximation in equation (4.1) is more valid for higher elements. Thus the change in orbital energies suggests that the heterolytic cleavage should be most di½cult for the CÐI bond, and the change in the interaction matrix element suggests the opposite. The interaction matrix element takes on double importance since the stabilization energy varies as the square of it. Whenever orbital interaction considerations involve opposing e¨ects, one must have recourse to experiment or higher level theory to elucidate the dominant factors. Evidently for halides, the change in DE het is dominated by the change in hAB, and heterolytic cleavage of the CÐI bond is easier than that of the CÐCl bond. The CÐBr bond is intermediate. The most di½cult to cleave heterolytically is the CÐF bond. One must bear in mind that heterolysis occurs only in polar media where additional bond forming (to solvent) takes place. Heterolysis of the CÐF bond is rarely observed. Likewise, the observed Lowry±Bronsted acidity of the HÐX acids is in the order HI > HBr > HCl > HF, as is consistent with the relative energies of the s orbitals or dominance of overlap considerations (Figure 4.3). Heterolytic cleavage of neutral molecules in the gas phase is never observed. The energy required to separate charges, of the order of the ionization potential of the H atom (13.6 eV, 1313 kJ/mol), is prohibitively high. However, it is readily compensated by solvation in polar solvents, especially water. Since solvation energies are very large, the simple theory proposed to this point could only rationalize gross trends. Heterolytic cleavage of charged species in mass spectrometric and negative-ion cyclotron resonance experiments is commonly observed.
Homolytic Cleavage of s Bonds Involving C or H
The energy change associated with homolytic bond dissociation is given by the equation (Figure 4.1)
|
hom |
ˆ ÿ2eL ‡ eA ‡ eB ˆ 2DeL ‡ DeAB A |
2hAB2 |
|
|
DE |
|
|
‡ DeAB |
…4:2† |
|
|
DeAB |
||||
Thus, the energy di¨erence of the ®nal singly occupied orbitals must also be considered. How would DE hom be expected to change in the series CÐC, CÐN, CÐO, and CÐF or the series HÐC, HÐN, HÐO, and HÐF? Recognizing, as before, that the orbitals of C, N, O, and F are all approximately the same size and therefore the interaction matrix element, hAB, will be approximately the same size for any A±B pair, the dominant factor determining the homolytic bond dissociation energy therefore is the di¨erence in orbital energies, which appears in the denominator of the ®rst term and as the second term. The two terms have the opposite e¨ect on the bond dissociation energy. To a ®rst approximation, the bond energies are expected to be similar if DeAB is small compared to jhABj, for example, CÐC and CÐN, and dominated by the second term if DeAB > jhABj, for example, CÐO and CÐF.
Similarly, one may ask how DE hom would be expected to change in the series CÐF, CÐCl, CÐBr, and CÐI or the series HF, HCl, HBr, and HI. The situation is simpler for these series, since both the electronegativity and orbital size vary but have the same e¨ect on the bond dissociation energy. Variation in orbital size is approximately pro-

76 SIGMA BONDS AND ORBITAL INTERACTION THEORY
TABLE 4.1. Average Homolytic Bond Dissociation Energies (kJ/mol) of RÐX
R |
X ˆ CH3 |
X ˆ NH2 |
X ˆ OH |
X ˆ F |
X ˆ Cl |
X ˆ Br |
X ˆ I |
H |
439 |
460 |
498 |
569 |
432 |
366 |
299 |
CH3 |
368 |
364 |
381 |
456 |
351 |
293 |
234 |
Source: CRC Handbook, CRC Press, Boca Raton, FL.
portional to n2, where n is the principal quantum number of the valence shell. The larger the discrepancy in orbital sizes, the smaller is the e¨ective overlap and the smaller is the interaction matrix element, hAB. It was argued above that for halides the ®rst term is dominated by the change in hAB. Since the electronegativity, and therefore DeAB, also decreases, one expects homolytic bond dissociation energies of the CÐX bonds to be in the order CÐI < CÐBr < CÐCl < CÐF [83]. Likewise, the observed strengths of the bonds of the HÐX acids are in the order HI < HBr < HCl < HF, as is consistent with the dominance of overlap considerations. Actual bond strengths at 298 K are shown in Table 4.1.
Homonuclear s Bonds CÐC, NÐN, OÐO, FÐF, ClÐCl, BrÐBr, and IÐI
Although our primary emphasis here is on s bonds to C and H, we will make a brief examination of the other ®rst-row homonuclear s bonds and the interhalogen bonds. Peroxides and the halogens are important reagents in organic chemistry. Orbital interaction theory will indicate how they react and why. The s bonds CÐC, NÐN, OÐO, and FÐF are compared in Figure 4.4. Following the principles expounded above, the energy changes as a consequence of the interaction, DeL and DeU, are the same for the homonuclear ®rst-row s bonds. The resulting orbitals are not shown; they are not polarized since the interacting orbitals are degenerate. The implication of constant DeL is that the homolytic bond dissociation energies (BDEs) are the same. This inference is
…a† |
…b† |
…c† |
…d† |
Figure 4.4. Homonuclear sXX bond and sXX antibond between atoms of the ®rst row: (a) X ˆ C; (b) X ˆ N; (c) X ˆ O; (d ) X ˆ F.

INTERACTIONS OF s BONDS |
77 |
…a† |
…b† |
…c† |
…d† |
Figure 4.5. Homonuclear sXX bond and sXX antibond between the halogens: (a) X ˆ F; (b) X ˆ Cl; (c) X ˆ Br; (d ) X ˆ I.
not quite justi®ed since each of the heteroatoms has occupied nonbonded orbitals which interact in a repulsive four-electron, two-orbital interaction. Conformational changes occur to minimize the repulsive interactions in the case of NÐN and OÐO. The twisted conformations of hydrazines and peroxides arise from this avoidance. In the case of peroxides, and especially halogens, signi®cant repulsive interactions remain and the OÐO and FÐF bonds are signi®cantly weaker.
The most dramatic observation in Figure 4.4 is the reduction in the energy of the s orbitals, the LUMO, in the series. As a consequence, the oxy and halo compounds are strong Lewis acids (subject to nucleophilic attack) and strong oxidizing agents (can readily accept electrons).
The interhalogen s bonds are compared in Figure 4.5. In this case, the intrinsic interaction matrix element decreases rapidly along the series F, Cl, Br, I, as does the electronegativity. The homolytic BDE will also decrease in the series, although not as fast since there will be less repulsion due to the lone pairs; because of the very small p-type overlap of n p orbitals …n > 2†, DeL A DeU. The opposite directions of eX and DeL suggest that the sXX orbital remains low and all of the halogens are good electrophiles and strong oxidants.
INTERACTIONS OF s BONDS
In our discussion of the interactions of s bonds, we will focus principally on bonds to carbon or to hydrogen or both. For most organic compounds (excluding those with three-membered rings, see below), the CÐC or CÐH s bonding orbital is too low in energy for the bond to function e¨ectively as an electron donor toward unoccupied orbitals of other molecules or other parts of the same molecule. Likewise, the s antibonding orbital is too high in energy to function as an electron acceptor from occupied orbitals of the main-group elements, either interor intramolecularly. A dramatic exception to this generalization is in their interaction with coordinatively unsaturated transition metal complexes, which may be simultaneously excellent donors and acceptors. The reactivity of s bonds will be discussed in greater detail in other chapters, but general principles can be expounded here.

78 SIGMA BONDS AND ORBITAL INTERACTION THEORY
…a† |
…b† |
…c† |
Figure 4.6. Diagrams for the interaction of two adjacent s bonds (only the two adjacent spn hybrid ends are shown): (a) perpendicular geometry; (b) anti coplanar; (c) syn coplanar.
There is ample evidence that s bonds do interact with other occupied orbitals and that s orbitals interact with other empty orbitals. Evidence for the latter case is primarily in the form of chemical activation of CÐH bonds by adjacent groups with low-lying empty or half-®lled orbitals. The increased acidity of CÐH bonds, that is, reactivity with Lewis bases, under these circumstances is discussed in Chapter 10. Several examples will serve to exemplify the interaction of s bonds with adjacent ®lled orbitals.
In the following discussion, the s bond will be represented by the sp3 hybrid orbital of the C atom. The important point to note is that this orbital is 75% p character and interacts with immediately adjacent neighbouring groups, including other s bonds, in a p fashion. Interaction of s bonds with neighboring s bonds depends on the orientation of one bond relative to the other (Figure 4.6). It is clear that the interaction is minimized (but not zero) when the two bonds are perpendicular to each other (Figure 4.6a) since then the adjacent spn hybrid orbitals lie approximately in each other's nodal planes. It is not immediately obvious, however, whether the coplanar anti (Figure 4.6b) or the coplanar syn (Figure 4.6c) arrangement results in maximum interaction. In fact, a large body of experimental data has been interpreted to show that the coplanar anti arrangement represents the strongest interaction between adjacent s bonds [6, 84]. Rates of electron transfer through chains of saturated CÐC bonds con®rm the anti con®guration is the most e¨ective for promoting through-bond coupling [85]. The interactions are repulsive in the four-electron case (s±s), attractive in the two-electron case (s±s ), as expected. It has been demonstrated that the attractive s±s interactions are more important than the repulsive s±s interactions and are responsible for the rotational barrier in ethane and ethanelike molecules, H3XÐYH3 (X,Y ˆ C, Si, Ge, Sn, Pb) [86].
As well as the relative orientation of the bonds, the extent of interaction will depend on the polarizations of the bonds. Bonds from C to an element less electronegative than C will have the larger coe½cient on C in the s orbital and smaller in the s orbital. Therefore, the two s bonds will experience a stronger interaction than the two s orbitals. This situation is depicted in Figure 4.7a. This is the case if the substituent is a metal.