

3 курс / Фармакология / Essential_Psychopharmacology_2nd_edition
.pdfCognitive Enhancers |
469 |
FIGURE 12 — 9. Acetylcholine (ACh) destruction and removal. Acetylcholine is destroyed by an enzyme called acetylcholinesterase (AChE), which turns ACh into inactive products. The actions of ACh can also be terminated by a presynaptic ACh transporter, which is similar to the transporters for other neurotransmitters already discussed earlier in relation to norepinephrine, dopamine, and serotonin neurons.
itory, and are blocked by atropine, scopolamine, and other well-known so-called anticholinergics discussed throughout this text (see, for example Fig. 11 — 11). Both nicotinic and muscarinic receptors have been further subdivided into numerous receptor subtypes, those for the muscarinic receptors being best known. Perhaps the Ml postsynaptic subtype of muscarinic receptor is the key receptor that mediates memory functions in cholinergic neurotransmission in cerebral cortical sites; however, a role for other cholinergic receptor subtypes has not been ruled out.
Cholinergic deficiency hypothesis of amnesia. Numerous investigators have shown that a deficiency in cholinergic functioning is linked to a disruption in memory, particularly short-term memory. For example, blockers of muscarinic cholinergic receptors (such as scolopamine) can produce a memory disturbance in normal human volunteers that has similarities to the memory disturbance in Alzheimer's disease. Boosr-ing cholinergic neurotransmission with cholinesterase inhibitors not only reverses

470 Essential Psychopharmacology
FIGURE 12 — 10. Acetylcholine (ACh) receptors. There are numerous receptors for ACh. The major subdivision is between nicotinic (N) and muscarinic (M) cholinergic receptors. There are also numerous subtypes of these receptors, best characterized for muscarinic receptor subtypes (M1, M2, Mx). Perhaps the M1 postsynaptic receptor is key to mediating the memory functions linked to cholinergic neurotransmission, but a role for other cholinergic receptor subtypes has not been ruled out.
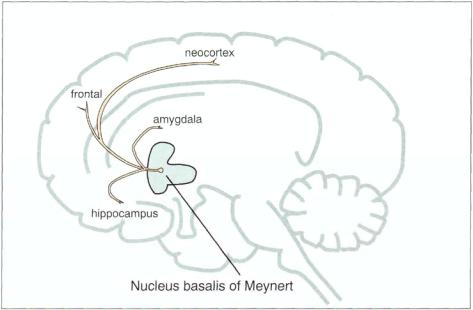
scopolamine-induced memory impairments in normal human volunteers but also enhances memory functioning in patients with Alzheimer's disease. Both animal and human studies have demonstrated that the nucleus basalis of Meynert is the major brain center for cholinergic neurons that project throughout the cortex. These neurons have the principal role in mediating memory formation (Fig. 12—11). It is suspected that the short-term memory disturbance of Alzheimer patients is due to degeneration of these particular cholinergic neurons. Other cholinergic neurons, such those in the striatum and those projecting from the lateral tegmental area (Fig. 12— 12), are not involved in the memory disorder of Alzheimer's disease.
Cognitive Enhancers |
471 |
FIGURE 12 — 1 1 . The nucleus basalis of Meynert, located in the basal forebrain, is the principal site of cholinergic cell bodies for axons that project to the hippocampus and amygdala and throughout the neocortex. These particular cholinergic neurons are thought to mediate memory and "higher" cortical functions, such as learning, problem solving, and judgment. They degenerate early and progressively throughout the course of Alzheimer's disease.
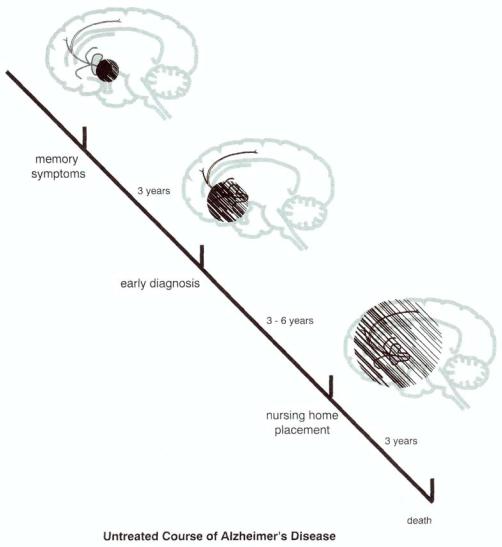
A "cholinergic deficiency syndrome" due to a limited degeneration of the nucleus basalis could theoretically also be responsible for the more limited short-term memory problems associated with "normal" aging (if there is such a thing), sometimes called mild cognitive impairment. Although Alzheimer's disease may start with a profound cholinergic deficiency, and this is the likely cause of memory disturbances early in its course, the illness is progressive, and many other symptoms develop, such as difficulties in problem solving, judgment, language, and behavior. Thus, it appears that degeneration begins at the nucleus basalis at the time of vague and undiagnosed memory symptoms (Fig. 12 — 13), spreading to closer projection areas such as the hippocampus, amygdala, and entorrhinal cortex by the time of early diagnosis, then spreading diffusely throughout the neocortex by the time of nursing home placement and loss of functional independence, and eventually involving many neurons and neurotransmitters systems by the time of death (Fig. 12
— 13). Here we will focus just on the cholinergic component of memory and just the memory component of Alzheimer's disease, recognizing that the whole picture is much more complex.
Impact of Memory Disorders on Cholinergic Neurotransmission
How Alzheimer's disease kills cholinergic and other neurons leading to memory loss. Alzheimer's disease is still a pathological, not a clinical diagnosis. It is thus defined by the presence of abnormal degenerative structures seen postmortem in the neocortex,

472 Essential Psychopharmacology
FIGURE 12 — 12. Other cholinergic neurons in the brain are thought to be involved in brain functions other than memory. They include interneurons in the striatum, which are involved in regulating motor movements, and those arising in the lateral tegmental area and projecting rostrally, caudally, and to the cerebellum with a wide variety of functions.
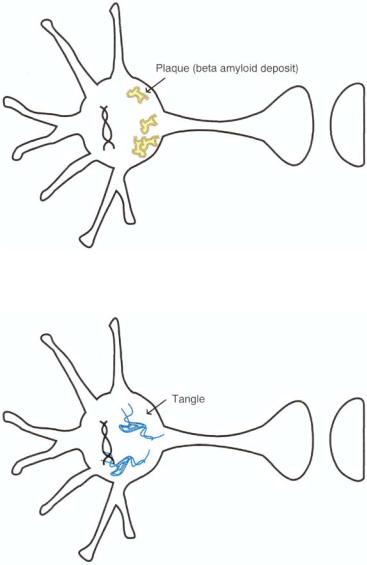
called neuritic plaques with beta-amyloid cores but also including other proteins such as apolipoprotein A (apo A) (Fig. 12 — 14) and neurofibrillary tangles of abnormally phosphorylated tau proteins (Fig. 12 — 15). Neuritic plaques are extracellular lesions, and their number correlate strongly with cognitive function. Presumably many are formed progressively in the cholinergic neurons of the nucleus basalis of Meynert (Fig. 12 — 13). Neurofibrillary tangles consist largely of a type of protein wrapped into bundles. These proteins are called tau proteins, which are chemically altered by being abnormally phosphorylated and then twisted together. The primary element of the neurofibrillary tangle is a paired helical filament, consisting of a ropelike section comprising two fibers wrapped around each other. It is hypothesized that these proteins interfere with nerve functioning in Alzheimer's disease, particularly in cholinergic neurons early in the course of illness, by impairing transport of molecules in the axon of these neurons. As time progresses, the tangles spread, and the consequences of deranged axoplasmic transport appear as a disorder of memory function as well as a disorder of other functions mediated by the cortex (Fig. 12 — 13).
Amyloid cascade hypothesis of Alzheimer's disease. A leading contemporary theory for the biological basis of Alzheimer's disease centers around the formation of beta amyloid. Certainly much of this amyloid destroys cholinergic neurons in the nucleus basalis of Meynert (Fig. 12 — 11), although the damage becomes more widespread as the disease progresses (Fig. 12 — 13). Hypothetically, Alzheimer's disease is a disorder in which beta amyloid deposition destroys neurons, in a manner somewhat analogous to that in which the abnormal deposition of cholesterol causes atherosclerosis. Thus,
Cognitive Enhancers |
473 |
FIGURE 12 — 13. The untreated course of Alzheimer's disease is progressive and downhill, beginning with very mild and nondiagnostic memory symptoms, probably signaling the beginning of a process in the nucleus basalis of Meynert. After about 3 years of nonspecific symptoms, a diagnosis of Alzheimer's disease is made, at which time damage to the cholinergic system has spread at least to the near projections from the nucleus basalis (i.e.., to the amygdala, hippocampus, and entorhinal cortex) and the person is losing a great deal of functional independence. In another 3 to 6 years, neurodegenerative progress now includes the neocortex diffusely; at this stage, the patient is in nursing home placement, and in a further 3 years is dead.
Alzheimer's disease may be essentially a problem of too much formation of beta amyloid, or too little removal of it.
One idea is that neurons in some patients destined to have Alzheimer's disease have an abnormality in the DNA that codes for a protein called amyloid precursor protein (APP) (Fig. 12—16). The abnormal DNA starts a lethal chemical cascade in neurons (Figs. 12 — 17 and 12 — 18), ultimately resulting in Alzheimer's disease (Figs.
474 Essential Psychopharmacology
FIGURE 12 — 14. Postmortem brain pathology defines what Alzheimer's disease is. Shown here are abnormal degenerative structures called neuritic plaques with amyloid cores.
FIGURE 12 — 15. Another key finding in Alzheimer's disease is the pathological finding of another degenerative structure called neurofibrillary tangles made up of abnormally phosphorylated tau proteins.
12 — 19 and 12 — 20). Specifically, the abnormal DNA causes the formation of an altered APP (Fig. 12 — 16), which instead of being removed from the neuron causes formation of beta amyloid deposits (Fig. 12 — 17). These deposits and fragments go on to form plaques and tangles (Fig. 12 — 18), the presence of which signals cell damage and cell death (Figs. 12-19 and 12 — 20). Sufficient cell damage and cell death give rise to the formation of the symptoms in Alzheimer's disease.
Another version of the amyloid cascade hypothesis is the possibility that something is wrong with a protein that binds to amyloid and removes it (Figs. 12
— 21

Cognitive Enhancers |
475 |
FIGURE 12—16. The amyloid cascade hypothesis of Alzheimer's disease (part 1). A leading contemporary theory for the biological basis of Alzheimer's disease centers around the formation of beta amyloid. Perhaps Alzheimer's disease is essentially a disease in which the abnormal deposition of beta amyloid reaches the point of destroying neurons. Thus, Alzheimer's disease may be essentially a problem of too much formation of beta amyloid, or too little removal of it. One idea is that neurons in some patients destined to have Alzheimer's disease have an abnormality in the DNA that codes for a protein called amyloid precursor protein (APP). The abnormal DNA starts a lethal chemical cascade in neurons, beginning with the formation of an altered APP.
FIGURE 12-17. The amyloid cascade hypothesis of Alzheimer's disease (part 2). Once altered amyloid precursor protein (APP) is formed (Fig. 12 — 16), it leads to the formation of beta amyloid deposits.
and 12 — 22). This protein is called APO-E. "Good" APO-E binds to beta amyloid and removes it, preventing the development of Alzheimer's disease and dementia (Fig. 12-21). In the case of "bad" APO-E, a genetic abnormality in APO-E formation causes it to be ineffective in binding beta amyloid. This results in beta amyloid deposition in neurons, which goes on to damage the neurons and cause Alzheimer's disease (Fig. 12 — 22).

476 Essential Psychopharmacology
FIGURE 12 — 18. The amyloid cascade hypothesis of Alzheimer's disease (part 3). Once beta amyloid deposits are formed from abnormal APP (amyloid precursor protein), the next step is that beta amyloid deposits form plaques and tangles in the neuron.
FIGURE 12-19. The amyloid cascade hypothesis of Alzheimer's disease (part 4). Formation of numerous neuritic plaques eventually causes the neuron to stop functioning and even to die.
Genes coding for APO-E are associated with different risks for Alzheimer's disease. There are three alleles (or copies) of the gene coding for this apolipoprotein which are called E2, E3, and E4. For example, a gene on chromosome 19 that codes for APO-E is linked to many cases of late-onset Alzheimer's disease. Moreover, APO-E is associated with cholesterol transport and involved with other neuronal functions, including repair, growth, and maintenance of myelin sheaths and cell membranes.

Cognitive Enhancers |
477 |
FIGURE 12-20. The amyloid cascade hypothesis of Alzheimer's disease (part 5). Formation of numerous neurofibrillary tangles also will eventually cause the neuron to cease to function and even to die.
FIGURE 12 — 21. Another version of the amyloid cascade hypothesis is the possibility that something is wrong with a protein that binds to amyloid and removes it. This protein is called APO-E. In the case of "good" APO-E, it binds to beta amyloid and removes it, preventing the development of Alzheimer's disease and dementia.
Having one or two copies of E4 increases the risk of getting Alzheimer's disease, and Alzheimer patients with E4 have more amyloid deposits.
Sporadic cases (i.e., noninherited cases) account for the vast majority of Alzheimer's disease cases, but about 10% of cases are inherited in an autosomal dominant

478 Essential Psychopharmacology
FIGURE 12 — 22. In comparison with Figure 12 — 21, where "good" APO-E can bind to beta amyloid and remove it, it is possible that patients with Alzheimer's disease have an abnormality in their DNA, which causes the formation of a defective or "bad" version of the APO-E protein. In this case, the bad APO-E cannot bind to beta amyloid, so the amyloid is not removed from the neuron.
Consequently, beta amyloid accumulates, forms plaques and tangles, and the neuron loses its function and dies.
fashion, and they have been intensely investigated for clues to the sporadic disorder. Such rare familial cases are also unusually early in their onset, and, unlike the common sporadic cases, have been linked to mutations in three different chromosomes, namely, 21, 14, and 1. The first mutation is on chromosome 21, where there is a defect in the gene for APP, leading to increased deposition of beta amyloid because a longer form of APP is formed. Recall that Down syndrome is a disorder of this same chromosome (i.e., trisomy 21), and virtually all persons with Down syndrome develop Alzheimer's disease if they live past age 50. The second mutation known to be associated with a familial form of early-onset Alzheimer's disease is on chromosome 14 in a gene called presenilin 1, which has no known relation to APP. The abnormal protein made by this gene mutation may have effects on membrane ion channels, intracellular protein transport, and cellular differentiation, all of which can affect the rate of neuronal degeneration. The third mutation known to be associated with a familial form of early-onset Alzheimer's disease is on chromosome 1, in a similar gene called presenilin 2, also unrelated to APP. It is not clear yet what, if anything, these three mutations in the rare familial cases tell us about the path-ophysiology of the usual sporadic, nonfamilial late-onset cases of Alzheimer's disease or, about how cholinergic neurons are damaged in them.
Other disorders that may kill cholinergic and other neurons, thus leading to memory loss.
Vascular dementia, formerly multi-infarct dementia, is characterized by dementia that classically has a more stepwise downhill course as compared with Alzheimer's disease, which has a more smoothly progressive downhill course. Multi-infact dementia is caused by multiple strokes, which damage the brain sufficiently to cause dementia and often cause focal neurological signs and symptoms as well. Normal pressure hydrocephalus can cause dementia from dilated cerebral ventricles. Creutzfeldt-