

6 курс / Неонатология / Пороки_развития_и_наследственные_заболевания_нервной_системы_у_детей
.pdfтазобедренных суставов: большой череп с маленькой лицевой частью, плоское лицо,
низко расположенные уши, запавшая переносица, гипеертелоризм, микрогнатия,
глоссоптоз, расщелина нёба, умственная отсталость, снижение слуха.
Описаны гидроцефалия, аринэнцефалия, лиссэнцефалия, гипоплазия или поликистоз почек, гидронефроз, гипоплазия лёгких, врожденные пороки сердца, гермафродитизм,
мужской кариотип у больных с женским фенотипом.
непропорциональная карликовость, искривление конечностей, гипоплазия лопаток,
плоское лицо, узкая грудная клетка.
Течение, Прогноз: Тип наследования- аутосомно-рецессивный.дети, как правило погибают в период новорождённости.Популяционная частота неизмвестна.
Соотношение полов М<Ж
Дифференциальный диагноз: другие формы врожденного искривления конечностей,
неонатальная форма несовершенного остеогенеза, гипофасфатазия.
Фанкони - Альбертини - Цельвегера синдром (s. Fanconi - Albertini - Zellweger),
псевдорахитическая ацидотическая остеопатия. Пероксисомные болезни.
Конституциональное нарушение обмена с ацидозом и остеопатией.
МКБ – 10: Q04.9 Врожденная аномалия мозга неуточненная
Этиология :Синдром наследуется по аутосомно-рецессивному типу, и, несмотря на то что заболевание является результатом единичных мутаций гена, при нем происходят многочисленные метаболические нарушения, в том числе обусловливающие
161
нарушение морфологического развития.Измененный фермент синдрома Цельвегера :
Пероксин 1, Пероксин 2, Пероксин 5, Пероксин 6
Патологическая анатомия: включает как нарушения нейро-онтогенеза, которые по данным аутопсии представлены пахимикрогирией, гетеротопиями или нарушениями процессов миелинизации, гипоплазией мозолистого тела, аномально сформированными обонятельными долями и нарушением образования борозд и извилин коры большого мозга. Наряду с этим отмечаются нарушение миелинизации вещества головного мозга, аномалии глаза. Патология внутренних органов включает гепатомегалию, фиброз печени, микроузелковый цирроз печени, холестаз. Биопсия печени выявляет депонированное железо и воспалительные изменения вокруг воротной вены. В почках имеются кисты коркового слоя. Сердечные аномалии встречаются часто: обнаруживаются дефекты межжелудочковой перегородки аномалии аорты.
Клиника : Синдром Цельвегера имеет характерный фенотип. Голова большая, как бы сдавлена с боков, с большим родничком и высоким лбом. Лицо необычное, с широким основанием носа, нос маленький, суженные носовые ходы, характерна гипоплазия надбровных дуг, постоянно открытый рот с выступающей нижней челюстью,
макроглоссия, микро- и гиподонтия, его углы загнуты, узкая щелевидная складка между веками, нерезкий гипертелоризм, множественные диастемы, дисплазия ушной раковины, шея короткая, хронический блефаро-конъюнктивит. Двусторонняя эквино-
варусная деформация стоп. Аномально сформированы половые органы – может быть крипторхизм, гипоспадия, клиторомегалия. В некоторых случаях возникает сложность клинически различить синдром Цельвегера и синдром Дауна. При рождении у детей с синдромом Цельвегера и синдром Дауна. При рождении у детей с синдромом Цельвегера бывает нормальная масса тела, но дети плохо поправляются и в дальнейшем отмечается задержка физического развития.
Неврологические нарушения включают судороги, резистентные к терапии, мышечную гипотонию. Офтальмологически выявляются пигментный ретинит, а также аномалии глаза, в том числе помутнение хрусталика (катаракта), глаукома, гипоплазия дисков зрительных нервов, что отражается аномальной электроретинограммой. Развивается
162
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
нейросенсорная глухота. Со стороны внутренних органов выявляются сердечные шумы из-за дефекта межжелудочковой перегородки и симптомы сердечной недостаточности, а также гепатоспленомегалия с высокими уровнями ферментов печени с дальнейшим развитием печеночной недостаточности и ДВС-синдрома.
Нарушение функции почек выражается в аминоацидурии и протеинурии. Из-за снижения функции надпочечников дети подвержены инфекционным заболеваниями.
Минимальные диагностические признаки : Пренатальная задержка роста, признаки лицевого дизморфизма, катаракта, пигментная ретинопатия, гепатомегалия,
умственная отсталость, поликистоз почек
Течение, Прогноз : Тип наследования, ген, синдрома цельвегера : АР, 7а 11.23, 8q21.1, 12, 6р21.1
Распространнённость синдрома Цельвегера 1 на 100 000
Прогноз: В течение заболевания могут появиться желудочно-кишечное кровотечение из-за коагулопатии, а также недостаточность надпочечников и остеопатия. В среднем смерть наступает в 12-13 нед от печёночной недостаточности и пневмонии.
Диагностика : Мр исследование часто выявляет нарушения миелинизации и неправильно сформированные борозды коры большого мозга. На рентгенограмме костей выявляется задержка костного возраста с типичным и точечными кальцификатами в надколенниках. Характерны гипопластическая анемия и повышение в плазме крови жирных кислот с очень длинной целью – это наиболее диагностически значимый признак при всех пероксисомных болезнях. В моче – альбумины,
лейкоциты, цилиндры, высокий уровень пипеколиновой кислоты.
Дополнительные скрининг-тесты обнаруживают низкий уровень плазминогена в культуре фибробластов, изменение каталазы от взвешенного состояния до выпадения осадка и дефицит пероксисомно-зависимых ферментов (дигидроацетон-фосфата,
ацетилтрансферазы и фитановой оксидазы).
Вызванные зрительные и слуховые потенциалы, Мр-исследование, КТ головного мозга, рентгенограмма скелета, анализ мочи, оценка ферментов печени и биопсия печени в совокупности позволяют установит диагноз с большой точностью.
Рентгенологически на фоне системного остеопороза отмечается недоразвитие ядер
163
окостенения, расширение ростковых зон, искривление метафизов и диафизов,
псевдопереломы.
В сыворотке крови обнаруживают уменьшение содержания белка, кальция, щелочных резервов.
В спинномозговой жидкости определяется повышенное содержание белка,
положительная реакция с коллоидным золотом.
Пренатальная диагностика с исследованием хориона или амнионической жидкости должна применяться в тех случаях, если у матери уже однажды родился ребёнок с пероксисомным заболеванием. Рсик повторного рождения составляет 25%.
Пренатальная диагностика основывается на определении уровня полиненасыщенных жирных кислот в амниотической жидкости. В случаях, если беременная женщина жалуется на уменьшение шевеления плода, то также можно заподозрить пероксисомное заболевание.
Дифференциальный диагноз : Периксисомное заболевания следует дифференцировать с глютаровой ацидемией, синдромом Прадера-Вилли и некоторыми врожденными миопатиями. Установить диагноз помогают дополнительные методы исследования.
Лечение не разработано. Применяют диету с ограничением жирных кислот с очень длинной цепью. Однако бесспорных доказательств улучшения состояния при этом получено не было.
COFS-синдром (cerebro-oculo-facio-skeletal syndrome), или церебро-окуло-фацио-
скелетный синдром Pena-Shokeir, type II
МКБ – 10: Q04.9 Врожденная аномалия мозга неуточненная
Клиника : Из пороков головного мозга при этом синдроме наиболее часто встречаются микроцефалия, иногда внутренняя гидроцефалия, атрофия зрительных нервов,
агенезия мозолистого тела, гипоплазия верхних теменных извилин, гиппокампа,
зрительных нервов, микрогирия базилярных поверхностей.
Отмечаются микрофтальмия, катаракта, уплощение передней камеры глаза, узкие глазные щели, блефарофимоз.
Аномалии лица: скошенный лоб, седловидная переносица, большие низко
164
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
расположенные уши, тонкие губы, ретро- и микрогнатия.
Из аномалий скелета встречаются кифоз, сколиоз, флексорные контрактуры суставов,
камптодактилия, стопа-качалка, вывих бедра, или дисплазия вертлужной впадины, coxa valga, узкий таз.
Со стороны внутренних органов чаще поражаются почки (агенезия и гипоплазия,
подковообразная почка), в единичных случаях отмечаются врожденные пороки сердца, релаксация диафрагмы, подвижная слепая кишка, гипоплазия селезенки. У
больных короткая шея, широко расставленные соски, поперечная ладонная складка,
мышечная гипотония, отставание физического и психомоторного развития
Минимальные диагностические признаки : резкая пренатальная гипоплазия,
микроцефалия, микрофтальмия или врождённая катаркта, скошенный лоб, тонкие губы, кифосколиоз, флексорные контрактуры суставов (артрогрипоз), задержка развития, постнатальная задержка роста .
Течение, Прогноз : Популяционная частота неизвестна. Соотношение полов - 1:1.
Тип наследования - аутосомно-рецессивный.
Тесты:
1)Укажите клинико-диагностические критерии синдрома Меккеля-
Грубера:
a)черепно-мозговая грыжа
b)поликистоз почек-увеличенный живот
c)стопа кочалка
d)поли-, син-, камптодактилия
e)флексорное положение конечностей
2) Укажите правильные ответы. Укажите характерную клиническую
картину при синдроме Ноя-Лаксовой.
a)Низкая масса тела и длины при рождении
b)стопа кочалка, узкий таз
c)ихтиозоформное изменение кожи
d)пороки развития глаз
165
e)контрактуры пальцев
3)Укажите соответствие между синдромами:
1.Смита-Лемми-Опитца
2.Кампомелическая дисплазия
a)короткий нос с открытыми вперед ноздрями
b)широкий альвеолярный край верхней челюсти
c)гипоплазия тел и отростков позвонков
d)укороченные конечности
Ситуационная задача: У ребенка при рождении низкая масса тела, микроцефалия,
узкий лоб, деормированные и низко расположенные ушные раковины, эпикант,
расходящееся косоглазие, короткий нос с широким кончиком и открытыми вперед ноздрями, широкий альвеолярный ркай верхней челюсти, флексорное положение пальцев рук. Ваш предварительный диагноз:
ГЛАВА 10
Синдром Апера (аутосомно-доминантный тип наследования); синдромы с неустановленным типом наследования: Гольденхара, Ланге, Рубенштейна-Тейби
Учебная цель: В процессе обучения магистрант должен изучать клинику,
диагностику синдромов Апера, Гольденхара, Ланге, Рубенштейна-Тейби.
Задачи: Магистрант должен знать:
1.Уметь дифференцировать синдромы Апера ,Гольденхара, Ланге,
Рубенштейна-Тейби.
2.Характерные признаки синдрома Апера ,Гольденхара, Ланге, Рубенштейна-
Тейби.
166
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
3.Клинические проявления синдромов Апера ,Гольденхара, Ланге, Рубенштейна-
Тейби.
1.Диагностику синдромов Апера ,Гольденхара, Ланге, Рубенштейна-Тейби.
Синдром Апера
МКБ-10. Q87.0
Синонимы: акрокраниодисфалангия, акросфеносиндактилия, акроцефалосиндактилия.
Этиология : За редкими исключениями синдром Апера вызывается одной из двух миссенс-мутаций гена FGFR2, вовлекающей две смежные аминокислоты: S252W и P253R, у 63% и 37% пациентов соответственно, по данным Wilkie и соавторов Синдактилия кистей и стоп была более серьезно выражена у пациентов с мутацией
P253R. Напротив, расщелины неба оказались более характерны для пациентов с мутацией S252W. Изучение фибробластов показало эктопическую экспрессию KGFR
области FGFR2, которая была связана с выраженностью патологий конечностей. Эта корреляция оказалась первым генетическим свидетельством того, что аномальная экспрессия KGFR является причиной синдактилии при синдроме Апера.
Клиника : Синдром Апера обычно диагностируется в раннем возрасте из-за обнаружения после рождения краниосиностоза и синдактилии. Для синдрома характерно наличие первичных изменений со стороны черепа уже при рождении,
однако окончательное формирование патологической формы происходит в течение первых трех лет жизни. У многих пациентов имеется затруднение носового дыхания,
из-за сокращения размера носоглотки и хоан, также могут быть затруднения прохождения воздуха через трахею, из-за врожденной аномалии хрящей трахеи, что может привести к ранней смерти. Возможны головная боль и рвота — признаки увеличенного внутричерепного давления, особенно в случаях, когда в процесс вовлечено несколько швов. Генеалогический анамнез представляется не столь важным, поскольку большинство случаев рождений детей с данным синдромом являются спорадическими.
167
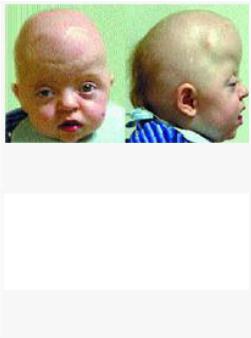
Фенотипические признаки синдрома Апера
Черепно-лицевая область. Наиболее часто встречается коронарный краниосиностоз, приводящий к акроцефалии, брахицефалии, туррибрахицефалии.
Синостозированию подвергаются также сагиттальные,
ламбдовидные, лобно-основные швы. Редкая аномалия черепа в виде трилистника найдена приблизительно у
 4% младенцев. Основание черепа уменьшено в
4% младенцев. Основание черепа уменьшено в
 размерах и часто асимметрично, передняя черепная
размерах и часто асимметрично, передняя черепная  ямка очень короткая. Передний и задний роднички увеличены в размерах и не заращены. Средняя линия свода черепа может иметь зияющий дефект,
ямка очень короткая. Передний и задний роднички увеличены в размерах и не заращены. Средняя линия свода черепа может иметь зияющий дефект,
простирающийся от области глабеллы через область метопического шва до переднего родничка, через область сагиттального шва до заднего родничка. Отмечаются: глазной гипертелоризм, экзорбитизм, мелкие орбиты, нависающие надбровные дуги. Со стороны глаз наблюдаются: экзофтальм, «прерывистые брови», пальпебральные трещины, косоглазие, амблиопия, атрофия зрительного нерва, и (редко) вывих глазного яблока, снижение пигмента, врожденная глаукома, обратимая потеря зрения.
Переносица часто запавшая. Нос короткий с уплощенной спинкой и с широким кончиком со стенозом или атрезией хоан, носогубные складки глубокие, возможна девиация носовой перегородки. Имеется гипоплазия средней зоны лица — верхняя челюсть гипоплазирована, скуловые дуги короткие, скуловые кости мелкие. В связи с этим отмечается относительный нижнечелюстной прогнатизм. Рот в состоянии покоя имеет трапециевидную форму. Высокое аркообразное небо, расщелина мягкого неба и язычка наблюдается в 30% случаев. Твердое небо короче, чем в норме, мягкое небо — длиннее и толще, верхнечелюстная зубная дуга имеет V-образную форму. Могут быть выступающие из ряда верхние зубы, имеющие форму совка резцы, сверхкомплектные зубы и выступающие альвеолярные гребни. Пациенты имеют низко посаженные уши и высокую вероятность снижения слуха в дальнейшем.
168
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
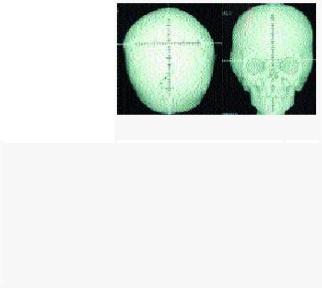
Конечности и скелет. Одним из основных проявлений синдрома является синдактилия кистей и стоп с вовлечением 2, 3 и 4-го пальцев. Реже в процесс вовлекаются 1-й и 5-
й пальцы. Проксимальные фаланги больших  Рисунок 2. Рентгенограмма
Рисунок 2. Рентгенограмма
пальцев кистей и стоп укорочены, дистальные
черепа больного с синдромом имеют трапециевидную форму. При синдроме Апера в прямой проекции
Апера центральные три пальца всегда
(формат 3 D)
подвергнуты синдактилии.
 При типе 1 — большой палец и часть 5-го пальца отделены от сросшихся пальцев;
При типе 1 — большой палец и часть 5-го пальца отделены от сросшихся пальцев;
 При типе 2 — только большой палец отделен от «среднего пальца»;
При типе 2 — только большой палец отделен от «среднего пальца»;
 При типе 3 — все пальцы сросшиеся.
При типе 3 — все пальцы сросшиеся.
Точно так же синдактилия пальцев стопы может вовлекать три боковых пальца (тип
1), или 2–5-й пальцы с отдельным большим пальцем ноги (тип 2), или может быть непрерывной (тип 3). При синдроме Апера патология верхних конечностей всегда более выражена, чем нижних. Сращение костей запястья с дистальными фалангами не имеет своего аналога на стопе. Возможны и другие патологические изменения конечностей: радиальное отклонение коротких и широких больших пальцев, из-за измененной проксимальной фаланги — брахидактилия; ограничение подвижности в плечевом суставе, ограниченная подвижность локтевого сустава с затруднением пронации и супинации, ограничение подвижности в коленном суставе, аплазия или анкилоз плечевого, локтевого и тазобедренного сустава. Одной из сравнительно часто встречаемых аномалий скелета при синдроме Апера является врожденное сращение позвонков. Наиболее характерно было сращение C5–C6. Сращение C5–C6 более характерно для синдрома Апера, а C2–C3 для синдрома Крузона, что помогает дифференцировать эти два заболевания. Рентгенографическое исследование шейного отдела позвоночника является обязательным перед анестезиологическим пособием для
169

этих пациентов. Прогрессивный синостоз отмечается не только в черепных швах, но и в костях ног, рук, запястьях, шейном отделе позвоночника и предложили термин
«прогрессирующий синостоз с синдактилией» как наиболее адекватно отражающий клиническую картину [24].
Кожа. По некоторым данным, для синдрома Апера характерны элементы глазо-кожного альбинизма (светлые волосы и бледная окраска кожных покровов). Наблюдается гипергидроз у всех пациентов. Также они описали акнеформные элементы, которые были особенно распространены на лице, груди, спине, руках. Помимо этого возможны проявления гипопигментации и гиперкератоза ладоней, западения кожи над крупными
суставами конечностей. У некоторых пациентов имеется избыточная кожа складок лба.
Центральная нервная система (ЦНС). С синдромом связаны различные степени умственного дефицита, однако есть сообщения и о больных с нормальным интеллектом. Повреждения ЦНС в большинстве случаев могут быть причиной умственной отсталости. Возможно, проведение краниоэктомии на ранних этапах способствует нормальному умственному развитию. Прогрессирующая гидроцефалия встречалась редко, и часто ее не удавалось дифференцировать с непрогрессирующей вентрикуломегалией.
Внутренние органы и системы. Для синдрома Апера характерны незначительные изменения со стороны внутренних органов. Патология со стороны сердечно-
сосудистой системы (дефект межжелудочковой перегородки, несращенный Баталлов проток, стеноз легочной артерии, коарктация аорты, декстракардия, тетрада Фалло,
эндрокардиальный фиброэластоз) отмечается у 10–20% больных. Аномалии мочеполовой системы (поликистоз почек, добавочные почечные лоханки,
гидронефроз, стеноз шейки мочевого пузыря, двурогая матка, атрезия влагалища,
увеличенные большие половые губы, клиторомегалия, крипторхизм) выявлены у 9,6%.
170
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/