

4. Алкилирование по атому азота (n-алкилирование)
В качестве алкилирующих агентов используют алкил- и арилгалогениды, непредельные соединения, спирты, эфиры, эпоксисоединения, диалкилсульфаты, эфиры аренсульфокислот. Поэтому чаще всегоN-алкилирование (арилирование) можно рассматривать как реакциинуклеофильного замещения (SN2, SNAr и др.) или нуклеофильного присоединения (AN). Особенности рассматриваемого процесса в большой степени зависят от используемого реагента.
Алкилирование аминов галогенидами идет по схеме:
![]()
Процесс сопровождается выделением галогеноводорода, который образует аммониевые соли и затрудняет реакцию, поэтому в реакционную массу добавляют вещества, связывающие кислоту. Ими могут быть сам амин, карбонаты натрия, калия, кальция или щелочь. Например, при полученииN-бензиланилина (производстводиазолина) для этих целей используют гидрокарбонат натрия:

Повышение нуклеофильности субстрата (амина, амида) существенно влияет на его активность и скорость реакции алкилирования. В данном случае нуклеофильность, а, следовательно, и активность субстрата, также, как и егоpKa, увеличивается от сульфамида до третичного амина:
–SO2NH2 < –CONH2 < ArNH2 < C5H5N < NH3 < RNH2 < R2NH < R3N
Алифатические амины и аммиак алкилируются легко, но процесс сопровождается полиалкилированием, что объясняется образованием более активного нуклеофила, чем исходный амин (см. главу 5 «Замена атома галогена на аминогруппы»):

Тем не менее, реакцию широко используют в химико-фармацевтической промышленности, т.к. многие лекарственные препараты выпускаются в виде четвертичных аммонийных солей, например,парамиони др.:
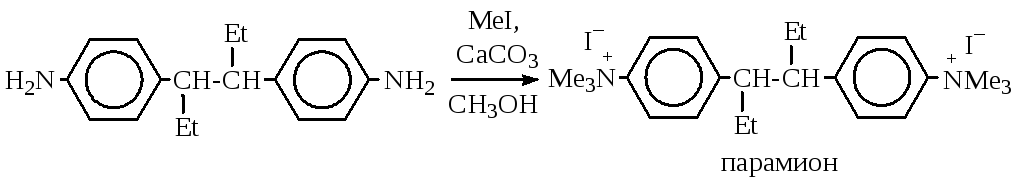
Алкилирование ароматических аминов идет труднее, чем алкиламинов, но селективнее. Это объясняется тем, что нуклеофильность их в большей степени зависит от заместителей в ядре, чем от заместителей, находящихся у атома азота. Так,п-толуидин более нуклеофилен, чем вторичный аминN-метиланилин:
![]()
Активность вторичного жирноароматического амина выше, чем ариламина, но не настолько, как в алифатическом ряду. Поэтому используя низкие температуры, избыток субстрата и другие приемы, можно получать смешанные аминосоединения с высоким выходом:
![]()
Амиды карбоновых и сульфоновых кислот, нуклеофильность которых очень низкая, алкилируются намного труднее, чем амины, но зато селективно, что позволяет синтезировать чистые первичные и вторичные амины (см. главу 5, «Замена атома галогена на аминогруппы»).
Алкилирующий агент также влияет на скорость реакции. Быстрее всего реагируют аллильные, бензильные, метильные и первичные галогениды (см. механизм SN2).
Как правило, алкилгалогенид является более простой молекулой, но в ряде случаев он является структурной основой лекарственного вещества, например, в производстве тримекаина:

При использовании полигалогенида можно избирательно заместить более хорошо уходящий или более активный галоген:
В производстве нейролептика метеразинаосуществляется замещение более хорошо уходящей группы (бромид иона) при алкилировании метилпиперазина 3-хлор-1-бромпропаном (в среде толуола в присутствии мелкоизмельченногоNaOHс азеотропной отгонкой воды и возвратом толуола):

Ниже приведены примеры замещения более активного галогена в синтезах антиаритмического препарата орнида:

Условия реакциизависят от строения и свойств как субстрата, так и галогенида. Так, при взаимодействии алкилгалогенидов с аминамитемпература реакции обычно до 100 °С. Активированные арилгалогениды в процессах арилирования реагируют с аминами примерно при 150 °С. В большинстве случаев реакцию можно вестипри атмосферном давлении в аппарате с обратным холодильником.
Однако при работе с низкокипящими веществами, такими, как метил- и этилхлориды (CH3Cl,C2H5Cl), которые значительно дешевле соответствующих бромидов и иодидов, алкилирование ведут вавтоклавах, что является основным недостатком этого метода. Реагенты обычно берут в стехиометрическом соотношении. Так, алкилирование анилина этилхлоридом до диэтиланилина ведут в присутствии мела (или извести) при 125 °С под давлением 1,0—1,2 МПа 12 часов.
![]()
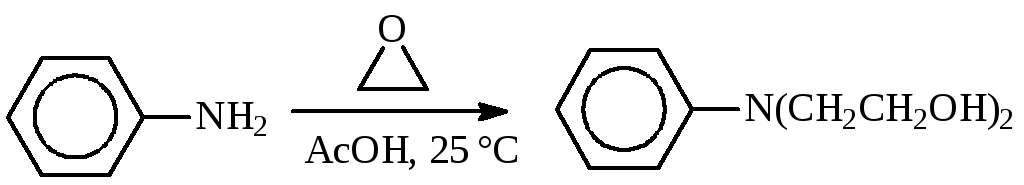
N-Алкилирование галогеноспиртами и эпоксисоединениями применяется в синтезе противоопухолевых препаратов. Для введения этанольного остатка в аминогруппу используют водный раствор этиленхлоргидрина (синтезыциклофосфана и проспидина):
![]()
![]()

Однако наряду с N-алкилированием идет и О-алкилирование.
Поэтому вместо этиленхлоргидрина часто используют окись этилена. Для получения монозамещенного производного реакцию ведут в большом избытке амина в присутствии воды. Для введения двух гидроксиэтильных остатков берут избыток окиси этилена и процесс проводят при небольшом давлении. Температура алкилирования обычно ниже 100 °С. Так, в синтезе хлорбутина реакцию гидроксиэтилирования проводят при температуре около 5 °С, сарколизина — не выше 25 °С:

Смесь окиси этилена с воздухом взрывается, поэтому алкилирование ведут при полном отсутствии воздуха, что достигается продувкой аппарата азотом.
Диметилсульфат применяется дляN-алкилирования в производстве целого ряда лекарственных препаратов. Ниже приводятся примеры использования его в синтезеазафена.

МеханизмреакцииSN2. В мягких условиях (водный раствор, низкая температура) используется лишьодна метильная группа диметилсульфата.Для полного использования диметилсульфата необходимо проводить алкилирование при температуре около 100 °С в щелочной среде.
К достоинствам диметилсульфата можно отнести высокую реакционную способность, относительную дешевизну и возможность проводить метилирование органических соединений, используя повышенные температуры при атмосферном давлении (для сравнения, температура кипения метилиодида ниже 40 °С). Серьезным недостатком диметилсульфата является его высокая токсичность.
Метиловые эфиры аренсульфокислот имеют достоинства диметилсульфата, но менее токсичны, поэтому их использование в качестве N-метилирующего средства в ряде случаев является более рациональным. Обычно процесс ведут начиная при 25—26 °С и заканчивая при 70 °С:

Непредельные соединения, взаимодействующие с аминами и азотистыми гетероциклами, также нередко используются в качестве алкилирующего агента в синтезе лекарственных соединений. Механизмреакции обычно рассматривают как нуклеофильное присоединение (AN) по-связи.
Так, в синтезе анатруксония пиперидин алкилируют малоактивным алкеном — аллиловым спиртом:
![]()
При получении диазолина используется активный метилакрилат, что позволяет при необходимости ввести в молекулу амина сразу два радикала:

Даже слабые нуклеофилы — амиды карбоновых кислот — алкилируются непредельными соединениями. Так, в синтезе пантотената кальция фталимид алкилируют акрилонитрилом в присутствии этилата натрия или 1 %-ного спиртового раствора едкого натра:

Метилирование аминов по Эшвайлеру-Кларку (формальдегидом в среде муравьиной кислоты) можно изобразить следующей схемой:


Процесс идет в два этапа. На первом образуются азометины или гидроксиметилпроизводные аминов, на втором продукты взаимодействия первичного и вторичного амина с формальдегидом восстанавливаются муравьиной кислотой до вторичного и третичного амина.
Выходпродукта высокий, иногда достигает 100 %. С помощью этого метода можно метилировать многие амины, в том числе, аминокислоты и гетероциклические амины при температурахоколо 100 °С.
Однако процесс алкилирования по Эшвайлеру-Кларку длительный, для его завершения требуется до 10—15 часов,«дорогой», используетсяагрессивнаясреда итоксичные вещества.Ароматические амины метилируются лишь при наличии орто- и пара-заместителей, препятствующих гидроксиметилированию ароматического ядра. В связи с этим он используется тогда, когда другие способы не дают хороших результатов, как правило, при наличии в молекуле нескольких нуклеофильных центров.
Этим методом метилируют 6-метилпиперидин-2-карбоновую кислоту (в производстве димеколина):

Во всех случаях выход продукта составляет около 90 %.
При этом метод позволяет целенаправленно, не затрагивая другие нуклеофильные центры, вводить в субстрат одну, две и более метильных групп. Например, при алкилировании 3-аминопропанола (синтез аминазина и пропазина):
![]()
При получении гидрохлорида диметилглицина (в синтезе витамина В15) метод Эшвайлера-Кларка также дает хороший выход, однако лучше идти другим путем, с использованием более дешевого и доступного сырья:

Замена формальдегида другими альдегидами и кетонами приводит к алкилированию аминов. Этот процесс обычно называют реакцией Лейкарта-Валлаха. В связи с тем, что карбонильные соединения и, особенно, кетоны менее реакционноспособны, чем формальдегид, алкилирование идет при более высоких температурах, чем метилирование.
Спирты в качестве N-алкилирующих агентовобычно используют в реакциях с ароматическими аминами.
В жидкой фазе процесс проводят в присутствии минеральных кислот в автоклавах под давлением выше 3 МПа, температуре 180—220 °С и в течение до 10 часов.
Так получают диметиланилиниз анилина, метилового спирта и серной кислоты (при использовании метилиодида — 125 °С, 1 МПа, 10 час).

В качестве побочного продукта образуется некоторое количество соли четвертичного аммониевого основания. Для разложения соли реакционную массу нагревают в автоклаве с раствором едкого натра.
Каталитическая роль кислоты заключается в протонировании спирта и образовании хорошо уходящей группы. Вода либо вытесняется нуклеофилом (SN2-механизм), либо отщепляется, образуя карбокатион, который реагирует с ароматическим амином (SN1-механизм):


Природа минеральной кислоты заметно влияет на скорость протекания реакции. Так, при алкилировании анилина избытком этилового спирта (под давлением при 180—200 °С) в присутствии соляной кислоты получается смесь продуктов, содержащая значительное количество моноэтиланилина. При использовании бромоводородной кислоты в тех же условиях образуется в основном диэтиланилин. Однако чаще всегоиспользуют более дешевые серную и соляную кислоты. Серную кислоту загружают из расчета до 0,3 моль, а соляную до 1 моль на моль амина.
Спиртдля алкилирования берется в избытке. При получении третичных аминов этотизбытокбольше (до 160 % от теории), при получении вторичных — меньше.
В паровой фазе алкилирование ароматических аминов спиртами проводят при температуре 300—400 °С в присутствииокиси алюминияв качестве катализатора.
В синтезах химико-фармацевтических препаратов алкилирование аминов спиртами имеет меньшее значение, чем другими реагентами. В качестве примера можно привести реакцию 1-фенил-2-пропанамина с гидроксиацетонитрилом в производствесиднофена:

N-Алкилирование простыми эфирами осуществляют в газовой фазе при температуре 250—350 °С. Смесь паров амина и эфира пропускают через слой катализатора (Al2O3,ThO2,TiO2,ZrO2).
Практический интерес представляет реакция анилина с метиловым эфиром, который является побочным продуктом в производстве метилового спирта:
![]()
В промышленной установке избыток паров метилового эфира смешивают в испарителе с парами анилина. Смесь паров поступает в контактный аппарат трубчатого типа, где на 94—96 % превращается в диметиланилин. После отделения метанола смесь аминов с метиловым эфиром поступает во второй контактный аппарат, после которого степень превращения анилина в диметиланилин достигает 99,5—99,6 % от теоретического. Общий выход диметиланилина с учетом потерь на других стадиях производства составляет 97,6 %. В качестве катализатора используется активированная окись алюминия. Катализатор работает без замены 5 лет. Этого удалось достичь благодаря применению испарителя с циркуляцией анилина при неполном его испарении. Установка производительностью 5000 тонн диметиланилина в год автоматизирована и обслуживается всего двумя рабочими в смену. Коррозия в производстве диметиланилина парофазным методом практически отсутствует, а потому вся аппаратура выполнена из обычной углеродистой стали.
5. О-Алкилирование (получение простых эфиров)
В качестве алкилирующих агентов гидроксигруппы могут быть использованы алкил(арил)галогениды, непредельные соединения, спирты, эфиры серной и сульфокислот.
Алкил(арил)галогениды широко применяются для О-алкилирования (арилирования). При этом используется реакция Вильямсона или реакция гидроксисоединений с галогенидами в присутствии щелочей:
![]()
![]()
Большой интерес представляют доступные и дешевые метил- и этилхлориды. Алкилирование ими ведут в автоклавах под давлением, поскольку эти вещества имеют низкую температуру кипения.
Так, метилирование гидрохинонапроходит при нагревании водного раствора его натриевой соли с хлористым метилом при температуре 100 °С и давлении 2 МПа:
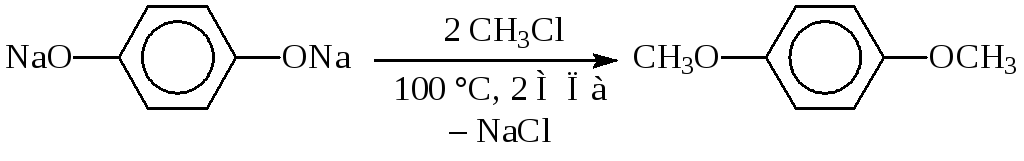
Галогенидычасто бываютсложными соединениями, а в ряде случаев их структура сложнее гидроксисоединения.
Примерами О-алкилирования (арилирования) могут быть:
Алкилирование фенола при получении нафтамонапроводят1,2-дибромэтаномв водно-щелочной среде при кипении реакционной массы:
![]()
При получении димедрола бензгидрол алкилируютβ-диметиламиноэтилхлоридом при 80 °С в щелочной среде:

п-Хлорфеноксиуксусную кислоту (в производствеацефена) синтезируют изп-хлорфенола ихлоруксусной кислотой в щелочной среде:

В производстве витамина В6 метиловый эфир метоксиуксусной кислоты получают, смешивая растворы метилата натрия (субстрата) ихлоруксусной кислоты (алкилирующего агента) в абсолютном метиловом спирте. В дальнейшем к реакционной массе добавляют концентрированную серную кислоту, кипятят 2 часа и отгоняют метиловый спирт. Выход продукта 70 %. При этом в реакциях алкилирования и ацилирования реагент сложнее, чем субстрат.
![]()
О-Алкилирование может быть использовано для временной защиты гидроксильных групп в гидроксилсодержащих соединениях. Одним из наиболее распространенных способов защиты являетсяметод тритилирования — образование трифенилметиловых (тритильных) эфиров. Чаще всего он применяется в синтезе сахаров, нуклеозидов и глицеридов.
Тритилгалогенид очень объемистая молекула, что значительно затрудняет его реакции с вторичными и третичными спиртами, поэтому метод тритилирования является эффективным способом блокирования первичных гидроксильных групп.
Реакцию проводят, нагревая спирт с рассчитанным количеством тритилхлорида в пиридине. С первичными спиртами она завершается за 1 час при 100 °С.
Тритиловые эфиры, как правило, легко кристаллизуются и обладают гидрофобностью, растворяясь в большинстве апротонных органических растворителей. Они устойчивы к действию щелочей и других нуклеофильных агентов, но гидролизуются в кислой среде. «Снимают» тритильную защиту, как правило, кипячением в 80 %-ой уксусной кислоте.

Таким образом, например, можно защитить первичные гидроксилы глицерина получив моно- и дитритиловые эфиры
Непредельные соединения для О-алкилирования используются реже. Так, при получении β-этоксипропионитрила (в синтезевитамина В1) используютакрилонитрил:
![]()
Процесс можно осуществлять также в пленочном реакторе непрерывного действия в присутствии 0,5—1 %-ного раствора этилата натрия.
Эту реакцию используют для модификации углеводов, например, в синтезе карбоксиэтил, карбамоилэтил и др. производных полисахаридов, где помимо акрилонитрила применяют акриламид:
![]()
О-Алкилирование спиртами в присутствии минеральной кислоты используется довольно редко и применяется, главным образом, для получения симметричных диалкиловых эфиров и алкоксипроизводных нафталинового и антраценового ряда.

О-Алкилирование эфирами серной кислоты и ароматических сульфокислот имеет значительно большее значение.
Реакция метилирования спиртов и фенолов диметилсульфатом в щелочной среде, также как и в случае аминов, протекает в две стадии. Щелочь повышает нуклеофильность субстрата и нейтрализует выделяющуюся кислоту:
![]()
![]()
Первая стадия идет легко при температуре ниже 50 °С, вторая — в гораздо более жестких условиях (примерно 100 °С) и часто проводится в автоклаве под небольшим давлением.
При метилировании неустойчивых природных соединений обычно используют лишь одну метильную группу диметилсульфата.
При метилировании фенолов при 100 °С метильные группы диметилсульфата используются примерно на 90 %, как, например, при алкилировании о-нитро-п-крезола:

Метилирование пирокатехина диметилсульфатом (в производствепапаверина) проводят в водном растворе NaOHпри 18—20 °С с последующей выдержкой при 90—92 °С:

Этим же способом можно метилировать одно- и многоатомные спирты, полисахариды.
Недостаткомметода является токсичность диметилсульфата и неполное использование метильных групп.
Алкилирование фенолов эфирами ароматических сульфокислот протекает гладко при кипячении с обратным холодильником смеси фенолята и соответствующего эфира аренсульфокислоты. В качестве растворителя используют полихлорбензол:
![]()