

V_N_Kovalev_Praktikum_po_farmakognozii
.pdfАнализ жирных масел |
7 1 |
|
|
|
|
|
|
|
Эфирное число — количество калия гидроксида, в миллиграммах, необходимое для омыления эфиров, содержащихся в 1 г исследуемого вещества.
Эфирное число IE вычисляют по формуле
IE = IS – IA,
где IS — число омыления; IA — кислотное число.
Йодное число — количество галогена в пересчете на йод, в граммах, которое присоединяется по месту двойных связей ненасыщенных жирных кислот в 100 г испытуемого вещества в описанных условиях. Йодное число показывает содержание ненасыщенных жирных кислот в 100 г жира.
Методика 1. Навеску вещества (в соответствии |
Т а б л и ц а 3.3 |
|
с табл. 3.3) помещают в сухую колбу с притертой |
||
Выбор навески |
||
пробкой вместимостью 250 мл, растворяют в 15 |
для определения йодного числа |
|
мл хлороформа, если нет других указаний в част- |
|
|
ной статье. К полученному раствору медленно при- |
|
|
бавляют 25 мл раствора йода бромида. |
|
|
Колбу закрывают пробкой и выдерживают |
|
|
в темном месте при частом перемешивании в те- |
|
|
чение 30 мин, если нет других указаний в частной |
|
|
статье. Прибавляют 10 мл раствора 100 г/л калия |
|
|
йодида, 100 мл воды и титруют раствором натрия |
|
|
тиосульфата 0,1 моль/л при интенсивном переме- |
|
|
шивании до светло-желтой окраски, затем при- |
|
бавляют 5 мл раствора крахмала и титруют раствором натрия тиосульфата 0,1 моль/л по каплям до обесцвечивания.
Параллельно проводят контрольный опыт. Йодное число II вычисляют по формуле
1,269 · (n2 – n1) II = ————————— ,
m
где n2 — количество раствора натрия тиосульфата 0,1 моль/л, израсходованное на титрование в испытуемом растворе, мл;
n1 — количество раствора натрия тиосульфата 0,1 моль/л, израсходованное на титрование в контрольном опыте, мл;
m — масса навески вещества, г.
Допускается проводить определение йодного числа по методике 2. Методика 2. Навеску испытуемого вещества (в соответствии с табл. 3.3)
помещают в сухую колбу с притертой пробкой вместимостью 250 мл, растворяют в 3 мл эфира, если нет других указаний в частных статьях, медленно прибавляют 20 мл раствора йода хлорида.
Колбу закрывают пробкой, смоченной раствором 10 г/л калия йодида, и выдерживают в темном месте при частом перемешивании в течение 1 ч, если нет других указаний в частной статье. Прибавляют 10 мл раствора 10 г/л калия йодида, 50 мл воды и титруют раствором натрия тиосульфата 0,1 моль/л при постоянном интенсивном перемешивании до светло-желтой окраски, затем прибавляют 3 мл эфира, интенсивно перемешивают, прибавляют 5 мл раствора крахмала и титруют раствором натрия тиосульфата 0,1 моль/л по каплям до обесцвечивания.
Параллельно проводят контрольный опыт. При анализе твердых жиров навеску растворяют в 6 мл эфира, прибавляют 20 мл раствора йода хлорида 0,1 моль/л. Дальнейшее определение проводят, как указано выше.
7 2 |
|
Тема 3. Липиды |
|
|
|
|
|
|
Полученные результаты сравнивают с данными табл. 3.1 и делают вывод о доброкачественности исследуемого образца жирного масла.
Гидроксильное число — количество миллиграммов калия гидроксида, эквивалентное количеству килоты, которое связывается при ацетилировании 1 г вещества. Методика определения гидроксильного числа изложена в теме «Эфирные масла».
Перекисное число — количество миллиэквивалентов активного кислорода, соответствующее количеству перекисей, содержащихся в 1000 г испытуемого вещества.
Если нет указаний в частной статье, используют метод А (ГФУ). Метод А. Около 5,0 г (точная навеска) вещества помещают в коническую
колбу с притертой стеклянной пробкой вместимостью 250 мл, прибавляют 30 мл смеси хлороформ— кислота уксусная ледяная (2:3). Колбу встряхивают до растворения вещества, прибавляют 0,5 мл насыщенного раствора калия йодида, перемешивают в течение 1 мин и прибавляют 30 мл воды. Полученный раствор титруют раствором натрия тиосульфата 0,01 моль/л, медленно добавляя титрант при непрерывном перемешивании почти до полного исчезновения желтой окраски. Затем прибавляют 5 мл раствора крахмала и продолжают титровать, интенсивно перемешивая до обесцвечивания раствора.
Параллельно проводят контрольный опыт.
Объем раствора натрия тиосульфата 0,01 моль/л, израсходованный на титрование в контрольном опыте, не должен превышать 0,1 мл.
Перекисное число IP рассчитывают по формуле
10 · (n1 – n2) IP = ————————,
m
где n1 — объем раствора натрия тиосульфата 0,1 моль/л, израсходованный на титрование исследуемого вещества, мл;
n2 — объем раствора натрия тиосульфата 0,1 моль/л, израсходованный на титрование в контрольном опыте, мл;
m — масса навески вещества, г.
Задание 7. Определите количество неомыляемых веществ в образце исследуемого масла. Произведите расчет и запишите результаты в лаборатор ный журнал.
Термин «неомыляемые вещества» применяется к веществам, нелетучим при температуре от 100 до 105 °С, которые экстрагируются органическим растворителем из испытуемого образца после его омыления. Содержание неомыляемых веществ вычисляется в % (мас. д.).
NB! Следует использовать стеклянную посуду со шлифами без смазки. Методика. Навеску испытуемого вещества, указанную в частной статье,
помещают в колбу вместимостью 250 мл, снабженную обратным холодильником. Прибавляют 50 мл раствора калия гидроксида спиртового 2 моль/л и нагревают на водяной бане в течение 1 ч, периодически перемешивая круговыми движениями. Затем охлаждают до температуры ниже 25 °С и содержимое колбы с помощью 100 мл воды переносят в делительную воронку. Полученный раствор осторожно встряхивают с тремя порциями эфира, свободного от пероксидов, по 100 мл каждая. Все эфирные извлечения собирают в другую делительную воронку, в которую предварительно помещают 40 мл воды, осторожно встряхивают в течение нескольких минут и оставляют до полного разделения слоев, после чего отбрасывают водный слой. Эфирный слой промывают двумя порциями воды, по 40 мл каждая. Затем тща-
Анализ жирных масел |
|
7 3 |
|
|
|
|
|
|
|
|
|
тельно отмывают |
поочередно 40 мл раствора 30 г/л калия гидроксида |
||
и 40 мл воды, повторяя данную процедуру 3 раза. Затем эфирный слой отмывают водой порциями по 40 мл до отсутствия щелочной реакции в водном слое по фенолфталеину. Эфирный слой количественно переносят в доведенную до постоянной массы колбу при помощи эфира, свободного от пероксидов.
Эфир отгоняют и к остатку прибавляют 6 мл ацетона. Растворитель тщательно удаляют в потоке воздуха. Остаток в колбе высушивают при температуре от 100 до 105 °С до постоянной массы, охлаждают в эксикаторе и взвешивают.
Содержание неомыляемых веществ, %, вычисляют по формуле
100 · а неомыляемые вещества = ————— ,
m
где а — масса остатка, г;
m — масса навески вещества, г.
Остаток растворяют в 20 мл спирта, предварительно нейтрализованного по фенолфталеину, и титруют спиртовым раствором натрия гидроксида 0,1 моль/л. Если израсходованный объем раствора натрия гидроксида спиртового 0,1 моль/л превышает 0,2 мл, разделение двух слоев было неполным; при этом взвешенный остаток не может рассматриваться как «неомыляемые вещества». В данном случае испытание следует повторить.
Задание 8. Проведите определение посторонних масел в образце жирного масла методом газовой хроматографии.
Испытание на посторонние масла проводят путем перевода жирных кислот, содержащихся в испытуемом масле, в метиловые эфиры.
Метод А (ГФУ). Этот метод неприменим для масел, содержащих глицериды жирных кислот с эпокси-, гидроксиэпокси-, циклопропиловыми или циклопропениловыми группами, а также для масел, в составе которых большая часть жирных кислот имеет длину цепи менее восьми атомов углерода и для масел с кислотным числом более 2,0.
Испытуемый раствор. Испытуемое масло высушивают перед метилированием, если это указано в частной статье. 1,0 г масла помещают в круглодонную колбу вместимостью 25 мл со шлифом, снабженную обратным холодильником и газоотводной трубкой. В колбу прибавляют 10 мл метанола безводного, 0,2 мл раствора 60 г/л калия гидроксида в метаноле, присоединяют обратный холодильник и, пропуская через смесь азот со скоростью около 50 мл/мин, встряхивают и нагревают до кипения. Когда раствор станет прозрачным (обычно через 10 мин), продолжают нагревание еще в течение 5 мин. Затем колбу охлаждают под проточной водой и содержимое переносят
вделительную воронку. Колбу промывают 10 мл гептана, переносят смывы
вту же делительную воронку и встряхивают. Прибавляют 10 мл раствора 200 г/л натрия хлорида и энергично встряхивают. Оставляют до расслоения, затем переносят органический слой в сосуд, содержащий натрия сульфат безводный, и через некоторое время фильтруют.
Раствор сравнения (а). Готовят 0,50 г смеси веществ, применяемых для калибровки (калибровочной смеси) состава, приведенного в одной из таблиц ГФУ, как указано в частной статье (если в частной статье не указан определенный раствор, готовят смесь состава, приведенного в табл. 3.4). Смесь растворяют в гептане и доводят объем раствора этим же растворителем до 50 мл.
Раствор сравнения (б). 1,0 мл раствора сравнения (а) доводят гептаном до 10,0 мл.
7 4 |
|
Тема 3. Липиды |
|
|
|
|
|
|
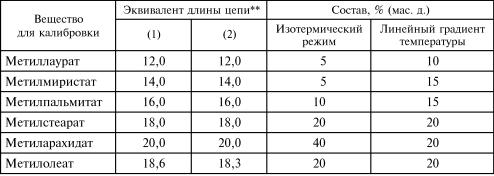
Т а б л и ц а 3.5
Приготовление смеси веществ, применяемых для калибровки*
П р и м е ч а н и е. * Для ГХ с применением капиллярной колонки и разделением потока рекомендуется добавлять к смеси веществ, применяемых для калибровки, компоненты с большей длиной цепи.
** Эти значения, вычисленные с применением калибровочной кривой, представлены как пример для колонки, заполненной полиэтиленгликольсукцинатом (1) и макроголом 20 000 (2).
Хроматографируют на газовом хроматографе с пламенно-ионизацион- ным детектором в следующих условиях:
—колонка стеклянная или из нержавеющей стали длиной от 2 до 3 м
ивнутренним диаметром от 2 до 4 мм, заполненная диатомитом для газовой хроматографии с размером частиц от 125 до 200 мкм, на который нанесено от 5 до 15 % полиэтиленгликольсукцината или полиэтиленгликольадипината;
—газ-носитель — азот для хроматографии;
—скорость газа-носителя — 25 мл/мин;
—температура колонки — 180 °С;
—температура устройства ввода проб и детектора — 200 °С.
При необходимости или если указано в частной статье, температуру колонки увеличивают от 120 до 200 °С со скоростью 5 °С в минуту.
Хроматографировать возможно также на газовом хроматографе с пламен- но-ионизационным детектором в следующих условиях:
—колонка капиллярная стеклянная или кварцевая длиной от 10 до 30 м
ивнутренним диаметром от 0,2 до 0,8 мм, внутренняя поверхность которой покрыта слоем поли[(цианопропил)(метил)][(фенил)(метил)]силоксана или макрогола 20 000 с толщиной слоя от 0,1 до 0,5 мкм или другой подходящей неподвижной фазой;
—газ-носитель — гелий для хроматографии или водород для хроматогра-
фии;
—скорость газа-носителя — 1,3 мл/мин (для колонки с внутренним диаметром 0,32 мм);
—деление потока — 1:100 или менее в зависимости от внутреннего диаметра применяемой колонки (в случае использования колонки с внутренним диаметром 0,32 мм деление потока должно составлять 1:50);
—температура колонки от 160 до 200 °С, в зависимости от длины колонки и используемой неподвижной фазы (для колонки длиной 30 м, покрытой слоем макрогола 20 000, температура должна составлять 200 °С);
—температура устройства ввода проб и детектора — 250 °С.
При необходимости или если указано в частной статье, температуру колонки увеличивают от 170 до 230 °С со скоростью 3 °С в мин (для колонки, покрытой слоем макрогола 20 000).
Анализ жирных масел |
7 5 |
|
|
|
|
|
|
|
Хроматографируют 0,5 мкл раствора сравнения (а). Чувствительность системы регулируют таким образом, чтобы высота основного пика на полученной хроматограмме составляла от 50 до 70 % шкалы регистрирующего устройства. Определяют времена удерживания жирных кислот, входящих в состав калибровочной смеси. Хроматографируют 1 мкл раствора сравнения (б) и рассчитывают отношение сигнал/шум для пика, соответствующего метилмиристату.
Хроматографируют от 0,5 до 1,0 мкл испытуемого раствора. Время хроматографирования должно в 2,5 раза превышать время удерживания метилолеата. Хроматограмму оценивают, как описано ниже.
При использовании калибровочных смесей № 1 или № 3 хроматографическая система считается пригодной, если выполняются следующие условия:
—на хроматограмме раствора сравнения (а) число теоретических тарелок n, вычисленное для пика, соответствующего метилстеарату, составляет не менее 2000 для набивной колонки и не менее 30 000 для капиллярной колонки;
—на хроматограмме раствора сравнения (a) коэффициент разделения
RS пиков, соответствующих метилолеату и метилстеарату, составляет не менее 1,25 для набивной колонки и не менее 1,8 для капиллярной колонки;
—на хроматограмме раствора сравнения (б) отношение сигнал/шум для пика метилмиристата составляет не менее 5.
При использовании калибровочной смеси № 2 хроматографическая система считается пригодной, если выполняются следующие условия:
—на хроматограмме раствора сравнения (а) число теоретических тарелок (n), вычисленное для пика, соответствующего метилкапрату, составляет не менее 1500 для набивной колонки и не менее 15 000 для капиллярной колонки;
—на хроматограмме раствора сравнения (a) коэффициент разделения Rs
пиков, соответствующих метилкаприлату и метилкапрату, составляет не менее 2 для набивной колонки и не менее 4 для капиллярной колонки;
— на хроматограмме раствора сравнения (б) отношение сигнал/шум для пика метилкапроата составляет не менее 5.
Оценка хроматограмм. Следует избегать условий хроматографирования, которые могут дать неразделенные пики (наличие компонентов с небольшим различием между временами удерживания, например кислоты линоленовая и арахидоновая).
Качественный анализ. Строят калибровочную кривую, используя хроматограммы раствора сравнения и данные табл. 3.5.
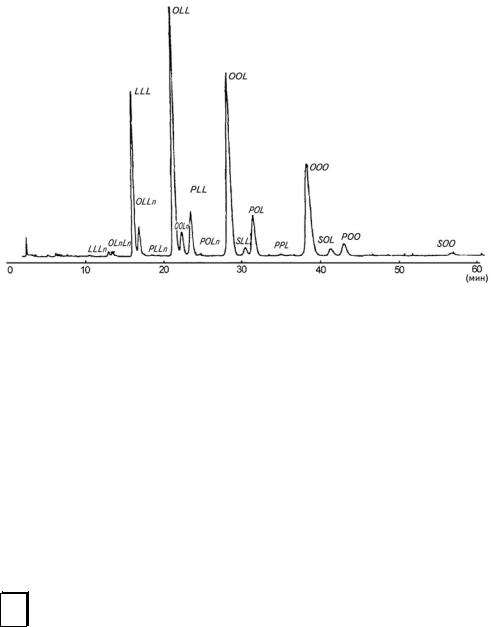
Пример типовой хроматограммы кунжутного масла (PhEur) приведен на рис. 3.4.
Для хроматограмм, полученных в изотермическом режиме, вычисляют логарифмы приведенных времен удерживания как функцию эквивалента числа атомов углерода в жирных кислотах. Калибровочная кривая насыщенных кислот представляет собой прямую линию. Логарифмы приведенных времен удерживания ненасыщенных кислот расположены на этой линии как точки, соответствующие нецелым значениям «эквивалента длины цепи». Идентификацию компонентов жирных кислот испытуемого масла проводят, рассчитывая логарифмы приведенных времен удерживания пиков, полученных на хроматограмме испытуемого раствора, и устанавливая по калибровочной кривой «эквиваленты длины цепи».
Для хроматограмм, полученных с использованием линейного градиента температуры, определяют времена удерживания, находящиеся в зависимос-
7 6 |
|
Тема 3. Липиды |
|
|
|
|
|
|
Рис. 3.4. Схема типовой хроматограммы кунжутного масла (PhEur):
Условия хроматографии: длина колонки 0,25 м; внутренний диаметр 4,6 мм; сорбент – октадецилсилил силикагель для хроматографии; подвижная фаза — смесь метиленхлорида и ацетонитрила (1:2).
Условные обозначения: Р — кислота пальмитиновая; S — кислота стеариновая; О — кислота олеиновая; L — кислота линолевая; Ln — кислота линоленовая
ти от числа атомов углерода в жирных кислотах, и идентифицируют жирные кислоты, входящие в состав испытуемого масла, путем сравнения с калибровочной кривой.
Количественный анализ. Обычно используют метод внутренней нормализации; при этом сумму площадей всех пиков на хроматограмме, кроме пиков, относящихся к растворителю, принимают за 100 %. Рекомендуется применение электронного интегратора. Содержание каждого компонента вычисляют как отношение площади соответствующего пика к сумме площадей всех пиков. Пики, площадь которых составляет менее 0,05 % от суммы площадей всех пиков, не учитывают, если нет других указаний в частной статье.
В определенных случаях, то есть при наличии жирных кислот с 12 или менее атомами углерода, в частной статье должен быть указан поправочный коэффициент для преобразования площадей пиков в проценты (м/м).
КОНТРОЛЬНЫЕ ВОПРОСЫ
?1. Дайте определение понятия «липиды»
2.Дайте определение понятия жиры (триацилглицериды).
3.Дайте определение понятия «липоиды».
4.Приведите типы классификации липидов.
5.Приведите классификацию жиров.
6.Приведите классификацию жирных масел.
7.Приведите общую формулу жира.
8.Охарактеризуйте кислоты, входящие в состав жиров и липоидов.
9.Перечислите способы получения жиров и жирных масел.
10.Охарактеризуйте метод количественного определения липидов в растительных объектах.

Анализ жирных масел |
7 7 |
|
|
|
|
|
|
|
11.Перечислите состав неомыляемого остатка жира.
12.Перечислите методы установления подлинности жиров (приведите примеры).
13.Охарактеризуйте физико-химические свойства жиров и жирных масел, физические и химические показатели жирных масел, их аналитическое значение.
14.Перечислите показатели доброкачественности жира.
15.Перечислите невысыхающие жирные масла.
16.Перечислите полувысыхающие жирные масла.
17.Перечислите высыхающие жирные масла.
18.Охарактеризуйте химический состав касторового масла, укажите его применение.
19.Охарактеризуйте химический состав льняного масла. Какое применение находит льняное масло?
20.Приведите пример твердого жира растительного происхождения, особенности его химической структуры и пути использования в медицинской практике.
21.Охарактеризуйте химический состав масла сои, укажите его применение и препараты.
22.Охарактеризуйте витамин F (фактор F), его биологическую активность и применение.
23.Приведите примеры жиров животного происхождения, которые используются в медицинской практике.
24.Приведите пример липоидов, их химический состав, способы получения и пути использования.
25.Какие соединения относятся к сопутствующим веществам жирных масел?

ФЕНОЛЬНЫЕ СОЕДИНЕНИЯ
К веществам фенольной природы принято относить ароматические соединения, которые содержат бензольное ядро с одной или несколькими гидроксильными группами.
В основу химической классификации фенольных соединений положен биогенетический принцип. Все фенолы можно разделить на несколько групп, расположив их в порядке усложнения молекулярной структуры, как показано в таблице.
Т а б л и ц а
Классификация фенольных соединений

ТЕМА
4Простые фенолы и их производные
В теме «Простые фенолы» объединены виды ЛРС, содержащие фенольные соединения с общей формулой С6, С6—С1 и С6—С2, как в гликозидированной форме, так и в виде фенолоальдегидов или фенолоспиртов, а также некоторые производные гидроксикоричных кислот. На рис. 4.1. представлены основные биологически активные производные простых фенолов.
Фенологликозиды — гликозидная форма соединений, у которых агликононом является фенильный радикал. Первый фенологликозид салицин, или О-β-D-глюкозид салицилового спирта, был выделен французским ученым Леру (1828) из коры ивы.
Рис. 4.1. Схема классификации некоторых фенолов
Анализ ЛРС, содержащего производные простых фенолов
Объекты для лабораторного исследования: листья толокнянки, листья брусники, кора ивы, корневища и корни родиолы розовой, корни и трава эхинацеи, трава фиалки, корневища папоротника мужского.
Объекты для самостоятельного изучения: листья черники, плоды ванили, корневища куркумы длинной, корни цикория, листья и корзинки артишока, корни лабазника вязолистного, корни и трава пиона уклоняющегося, трава конопли, плоды малотуса (камалы), кора корней хлопчатника.

8 0 |
|
Фенольные соединения. Тема 4. Простые фенолы и их производные |
|
|
|
|
|
|
ЛИСТЬЯ ТОЛОКНЯНКИ — Folia Uvae ursi
Задание 1. Изучите по гербарному образцу и рис. 4.2 толокнянку. Запиши те в лабораторный журнал название сырья, лекарственного растения и семей ства на русском и латинском языках. Поясните происхождение народного назва ния «медвежье ушко»? Обратите внимание на срок заготовки сырья.
Задание 2. Проведите анализ листьев толокнянки в сравнении со стандарт ным образцом сырья. Запишите, используя схему 7, основные внешние признаки исследуемого сырья. Обратите внимание на жилкование и форму верхушки листа (цв. вкл. II, рис. 1).
Внешние признаки по ст. 26 ГФ XI. Листья мелкие, кожистые, плотные, ломкие, цельнокрайние, обратнояйцевидной или удлиненно-овальной формы, на верхушке закругленные, иногда с небольшой выемкой, к основанию клиновидно суженные, с очень коротким черешком. Длина листа 1—2,2 см, ширина 0,5—1,2 см. Жилкование сетчатое.
Листья с верхней стороны темно-зеленые, блестящие, с ясно заметными вдавленными жилками, с нижней стороны немного светлее, матовые, голые.
Запах отсутствует. Вкус сильно вяжущий, горьковатый.
Рис. 4.2. Толокнянка обыкновенная (медвежье ушко):
а — побег; б — лист (вид сверху)
Задание 3. Приготовьте микро препарат листа толокнянки с поверх ности и рассмотрите его при м/у и б/у и зарисуйте в лабораторном журнале основные диагностические признаки (рис. 4.3).
Задание 4. Приготовьте извлече ние из образца исследуемого сырья и проведите качественные реакции на арбутин и дубильные вещества. На ос новании результатов проведенных ре акций сделайте заключение о химиче ском составе листьев толокнянки. Поясните, почему не проводится стан дартизация ЛРС по содержанию ду бильных веществ.
Методика. Измельченные листья толокнянки (масса навески 0,5 г) кипятят с 10 мл воды в течение 2—3 мин. Раствор фильтруют горячим через бумажный фильтр. Фильтрат используют для проведения качественных реакций.