

1211
.pdfаб
Рис. 5. Деформированный образец с решеткой графена: а – неоднородная решетка при однородном деформировании; б – однородная решетка при дополнительном относительном смещении (3)
После наложения смещений (3) на слой B получается однородное распределение атомов в деформированной решетке графена
(рис. 5, б).
Заметим, что при растяжении-сжатии вдоль оси Ox3 однородность строения кристалла гексагонального графита не нарушается и дополнительного смещения системы атомов B относительно системы атомов A не требуется.
6.2.2. Основы дискретного подхода
6.2.2.1. Метод молекулярной динамики
Появление вычислительной техники позволило рассмотреть точные выражения для сумм сил взаимодействия атомов по различные стороны от произвольной площадки в твердом теле при вычислении напряжений. При этом не требуется накладывать никаких упрощающих предположений относительно распределения частиц в теле и линеаризации взаимодействия, но необходимо задавать конкретный вид силы взаимодействия и значений всех ее параметров.
В нашей стране одним из первых стал применять динамические расчеты движения и взаимодействия атомов и молекул различных
21
конденсированных сред, в том числе жидкостей, вблизи поверхностей раздела фаз Аллан Георгиевич Гривцов [17]. Математическая постановка задач молекулярной динамики основана на уравнениях классической механики с конкретизацией сил межчастичного взаимодействия. Для этих сил делается предположение, как и в статистической механике, об их консервативности. Тогда вопрос о математическом описании конкретного типа взаимодействия частиц сводится к выбору его потенциала. Уравнения движения имеют вид [18]
|
mrk = |
N |
|
N |
||||
|
∑F(rki )rki +∑Ψ(rki ,vki )rki +φ(rk ) + ψ(rk ,vk ) , |
|||||||
|
&& |
|
|
|
|
|
||
|
|
|
|
|
|
i=1 |
i=1 |
|
где m – масса частицы; |
rki |
≡ rk −ri – разность радиус-векторов час- |
||||||
тиц; |
rki = |
|
rki |
|
, |
vki ≡ vk |
−vi |
– разность скоростей частиц; члены |
|
|
|||||||
F(rki ) |
и Ψ(rki ,vki ) описывают консервативную и неконсервативную |
|||||||
составляющие силы взаимодействия между частицами; векторзначные функции φ(rk ) и ψ(rk ,vk ) описывают внешнее консервативное
и неконсервативное силовые поля соответственно. Приняты обозначения F(r) = f (r) / r , f (r) = −Φ'(r) , где f (r) – скалярная сила взаи-
модействия частиц, Φ(r) – потенциал взаимодействия. Неконсервативное взаимодействие Ψ(rki ,vki ) предназначено
для описания внутренней диссипации в теле. Внешние силовые поля φ(rk ) и ψ(rk ,vk ) обычно используются для двух целей – для зада-
ния внешних массовых силовых воздействий (гравитационного, электромагнитного) и для задания силовых граничных условий. В 1-м случае эти силы распределены во всем объеме пространства, где проводится расчет, во 2-м случае они локализованы вблизи некоторых поверхностей, часто являющихся границами расчетной области. Кроме того, неконсервативное воздействие ψ(rk ,vk ) часто
используют для описания отвода энергии из системы посредством внешней диссипации, простейшим вариантом которой является сила вязкого трения ψ(rk ,vk ) = −Bvk , B > 0 . Эта сила также использует-
22
ся для внешнего контроля над тепловым движением атомов в системе. В этом случае коэффициент B может быть знакопеременным
изависеть от уровня тепловой энергии всей системы.
Впростейшем случае в отсутствие неконсервативного взаимодействия и внешних сил получим уравнение движения вида
mxi = ∑f ( |
xk −xi |
)(xk −xi ) / |
xk −xi |
. |
(4) |
&& |
|
|
|
|
|
k |
|
||||
Приведем основные ограничения на функцию, задающую потенциал. Для любых двух частиц, находящихся бесконечно близко
друг к другу, сила отталкивания бесконечно велика lim f (r) = +∞.
r→0
На бесконечности сила ничтожно мала, причем в расчетах часто принимают, что уже при расстояниях между частицами более 2a, где a – равновесное расстояние для пары атомов, обеспечиваемое этим
потенциалом, сила приближенно считается нулевой lim f (r) = 0 .
r→∞
Система (4) представляет собой задачу Коши, начальными условиями для которой являются распределения частиц в пространстве rk и их скоростей vk . Эти распределения могут быть самыми
разными в зависимости от целей исследования, но обычно для исключения движения рассматриваемой системы частиц как жесткого целого должны выполняться требования:
N |
N |
∑mk vk = 0 , ∑mk (rk −r0 )vk = 0 . |
|
k =1 |
k =1 |
ПотенциалЛеннарда-Джонса. Двухчастичный потенциал Лен- нарда-Джонса и соответствующая сила взаимодействия имеют вид
|
12 |
α |
6 |
|
12β |
|
13 |
α |
7 |
|
|
||
|
α |
|
|
|
α |
|
|
|
|||||
Φ(r) =β r |
−2 r |
|
, f (r) = |
|
r |
− r |
|
, |
(5) |
||||
|
α |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
где β – энергия связи, α – равновесное расстояние для пары атомов. Потенциал Леннарда-Джонса является двухпараметрическим и весьма
23
точно описывает свойства ряда веществ (прежде всего, кристаллических инертных газов), а также достаточно точно описывает силы взаимодействия Ван-дер-Ваальса. К несомненному достоинству потенциала Леннарда-Джонса относится также его вычислительная простота, не требующая вычисления иррациональных и трансцендентных функций. Потенциал Леннарда-Джонса широко применяется как классический модельный потенциал.
Потенциал Ми. Потенциал Ми и соответствующая ему сила межатомного взаимодействия имеют вид
|
β |
|
|
α |
n |
α |
m |
|
nm β |
|
α |
n+1 |
α |
m+1 |
|
||
|
|
|
|
f (r) = |
|
|
|
|
|||||||||
Φ(r) = |
|
m r |
|
−n r |
, |
|
|
|
r |
|
− r |
|
. |
||||
n −m |
|
n −m |
α |
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Потенциал Ми является четырехпараметрическим, что дает большую свободу при выборе параметров.
Потенциал Морзе. Потенциал и сила взаимодействия Морзе имеют вид
Φ(r)=β e−2γ(r−α) −2e−γ(r−α) , f (r)= 2αD e−2α(r−α) −e−α(r−α) .
Потенциал Морзе является трехпараметрическим и применяется в расчетах многими исследователями. Его достоинством по сравнению с потенциалом Леннарда-Джонса является более быстрое затухание на расстоянии, что удобно, если при моделировании необходимо учитывать взаимодействие только ближайших частиц. Недостатком потенциала Морзе является необходимость вычисления экспоненты, что приводит к замедлению расчетов.
Модифицированный потенциал. Часто возникает необходи-
мость изменить степень дальнодействия потенциала, сохранив его основные свойства. Для этого используется следующий модифицированный потенциал:
Φ% (r) = Φ(k(r −α) +α),
где Φ – некоторый произвольный потенциал взаимодействия. При k =1 исходный и модифицированный потенциалы совпадают, при
24
k >1 модифицированный потенциал «сжат» по сравнению с исходным, при k <1 он «растянут». Отметим, что Φ% (α) = Φ(α) , следова-
тельно, «сжатие» и «растяжение» происходит относительно точки равновесия r = α . Тогда сила взаимодействия
f%(r) = k f (k(r −α) +α) .
Отметим, что модифицированный потенциал, полученный из потенциала Морзе, является потенциалом Морзе с измененным значением γ% = k γ . Модифицированный потенциал, полученный из по-
тенциала Леннарда-Джонса при k = γ 6 , эквивалентен потенциалу Морзе, определяемому параметром γ . Под эквивалентностью будем понимать возможность совпадения у потенциалов трех размерных параметров: равновесного расстояния α, жесткости связи и энергии связи β. Таким образом, использование модифицированного потенциала позволяет расширить взаимодействие Леннарда-Джонса, сделав его трехпараметрическим по аналогии с потенциалом Морзе.
6 , эквивалентен потенциалу Морзе, определяемому параметром γ . Под эквивалентностью будем понимать возможность совпадения у потенциалов трех размерных параметров: равновесного расстояния α, жесткости связи и энергии связи β. Таким образом, использование модифицированного потенциала позволяет расширить взаимодействие Леннарда-Джонса, сделав его трехпараметрическим по аналогии с потенциалом Морзе.
Более подробное описание различных потенциалов приводится в работах [17, 18]. Здесь же еще раз подчеркнем, что на качественном уровне практически все потенциалы дают одинаковое описание межатомного взаимодействия.
При исследовании механических свойств различных тел методом молекулярной динамики для построения равновесных кривых зависимости физико-механических свойств исследуемых материалов, например зависимости напряжений от деформаций при определении упругих модулей, требуется подводить к системе атомов механическую энергию. Эта энергия может подводиться либо через задание перемещений граней образца, либо через приложение к граням некоторых сил. Подведенная механическая энергия перераспределяется по образцу и в энергию деформирования, и в энергию теплового движения атомов. Контролировать процесс распределения энергии невозможно и отделить чисто упругое поведение образца от термоупругого также не представляется возможным. Кроме того, динамические методы позволяют работать с небольшим числом час-
25
тиц на сравнительно малых временах (наносекунды). Прикладываемые при этом внешние воздействия достигают конечных значений за еще меньшее время, т.е., по сути, являются примерами высокоскоростных (ударных) нагрузок. Определяемые при таких воздействиях упругие модули являются адиабатическими, существенно превышающими известные в справочной литературе изотермические (статические) упругие модули. Таким образом, при динамическом моделировании после подвода к системе частиц механической энергии необходимо проводить расчеты по переходу системы в равновесное состояние и затем определять значение требуемого свойства в равновесном состоянии. При этом желательно для каждой точки получаемой зависимости проводить осреднение по достаточной для статистической обработки выборке реализаций рассматриваемой системы частиц. Всеэто требует огромного вычислительного времени.
6.2.2.2. Метод атомарной статики
Прогнозирование упругих модулей кристаллических материалов на основе углерода предлагается осуществлять с использованием статического подхода [19, 20]. При моделировании ковалентной связи в графене считается, что составляющая потенциала взаимодействия, связанная с отталкиванием атомов, действует между всеми атомами образца, а притяжение – только для ближайших атомов, расположенных в направлении действия связи согласно структуре sp2-гибридизированной электронной оболочки. При этом при построении компьютерной модели мы уходим от вопроса об устойчивости решетки или изменения направления ковалентной связи при деформировании решетки графена.
Принимается, что для определения величины и направления силы взаимодействия атомов может использоваться любой двухчастичный потенциал. Качественно картина всегда одинакова: если два атома участвуют в силовом взаимодействии, то при сближении атомы отталкиваются, а при удалении притягиваются, вне зависимости от вида связи. В статическом подходе принимается, что направление связи уже учтено и соответствующая ему структура кристалла задана, а по ней определяются ее упругие свойства.
26
Для описания взаимодействия атомов далее выбирается степенной потенциал Ми или потенциала Леннарда-Джонса. Это обосновано тем, что при расчете упругих модулей нет необходимости рассматривать динамические процессы, идущие при сверхнизких температурах или при скоростном деформировании, когда важную роль во взаимодействии атомов могут играть квантовые эффекты, учитываемые в потенциалах специального вида. Использование статической постановки и степенных потенциалов позволяет получить аналитическое решение задачи определения равновесного межатомного расстояния для различных кристаллических решеток и образцов различного размера. При этом точные значения межатомного расстояния получаются для объемов материала с небольшим числом атомов N на ребре образца (от 5 до 50). Далее для получения макроскопических значений параметров по этим точным решениям можно сделать предельный переход, устремив число атомов на ребре образца к бесконечности. В частности, это позволяет проводить идентификацию параметров потенциала по известным макроскопическим физико-механическим свойствам материалов.
6.2.3.Введение температуры
Впредыдущих подразделах не обсуждался вопрос об учете температуры при расчете упругих модулей монокристаллических твердых тел. Рассмотрим способ задания температуры в статическом подходе. При моделировании поведения механических свойств различных тел методом молекулярной динамики температура вводится через кинетическую энергию движения атомов относительно среднего движения тела. При стабилизации системы энергия крупномасштабного движения атомов переходит в мелкомасштабные колебания с меньшей амплитудой. Эти мелкомасштабные колебания относительно не меняющихся со временем центров отождествляются
степловым движением, и с помощью выражения E = kT , где E – амплитуда пульсаций кинетической энергии, k – постоянная Больцмана, определяется температура тела T. При построении равновесных кривых зависимости физико-механических свойств исследуемых
27
материалов (например, коэффициента теплового расширения, внутренней энергии, упругих модулей) от температуры при динамическом моделировании после подвода к системе частиц энергии, вызывающей повышение температуры, необходимо проводить расчеты по переходу системы в равновесное состояние и затем определять значение требуемого свойства в равновесном состоянии. При этом желательно для каждой точки получаемой зависимости проводить осреднение по достаточной для статистической обработки выборке реализаций рассматриваемой системы частиц. В статическом подходе нет возможности вводить температуру как характеристику теплового движения атомов, поскольку само движение при этом исключается. Можно было бы считать, что статический подход дает возможность определять равновесные положения центров, относительно которых атомы совершают колебательное движение, не зависимое от перемещений этих центров, связанных с механическим движением как жесткого целого или с деформированием тела. Тогда температуру в статическом подходе можно учесть с помощью введения некоторой искусственной дополнительной силы, приводящей к увеличению при нагреве межатомного расстояния в равновесной конфигурации, свободной от внешних механических воздействий.
Для выполнения индивидуальной научно-исследовательской работы магистрантам предлагается другой путь – имитация тепловых колебаний атомов с некоторой заданной амплитудой. Последнее достигается наложением на систему атомов случайных равновероятных по направлениям в пространстве смещений с этой амплитудой. Частота этих смещений (колебаний) в статическом подходе рассматривается как независимый параметр, требующий идентификации для конкретного материала наравне с параметрами потенциала. Для корректного задания тепловых возмущений атомов необходимо гарантировать равномерное распределение направления случайных смещений в пространстве. Направление будем задавать единичным вектором, поэтому задача о выборе направления случайного смещения атома в некоторый момент его теплового движения сводится к нахождению равномерного распределения точек насфере единичного радиуса.
28
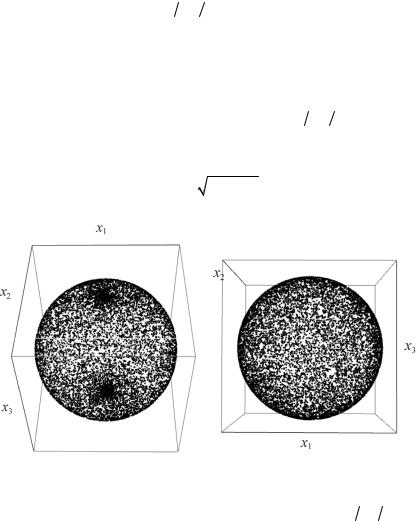
Алгоритм получения равномерного распределения точек по сфере. В сферической системе координат равномерное разбиение интервалов для угла ϕ [0;2π) , откладываемого от оси Ox1 в плос-
кости x1Ox2, и угла θ [−π 2;π 2], откладываемого от плоскости
x1Ox2, приводит к концентрации точек на полюсах сферы (рис. 6, а). Связь декартовых и сферических координат имеет вид x1 = rcosθcosϕ, x2 = rcosθsinϕ, x3 = rsinθ. Для того чтобы не про-
исходило концентрации точек вблизи полюсов сферы θ= ±π 2 , разбивать необходимо не интервал для угла θ [−π 2;π 2], а интервал [−1;1] значений функции sinθ. Функция cosθ (положительная для рассматриваемого интервала значений угла) выражается через полу-
2 , разбивать необходимо не интервал для угла θ [−π 2;π 2], а интервал [−1;1] значений функции sinθ. Функция cosθ (положительная для рассматриваемого интервала значений угла) выражается через полу-
ченное случайное значение как 1−sin 2 θ . В результате получается равномерное покрытие сферы точками (рис. 6, б).
а |
б |
Рис. 6. Виды распределений точек по единичной сфере, полученные:
а– при равномерном разбиении интервала для угла θ [−π 2;π 2] ,
б– приравномерномразбиении интервала [−1;1] значенийфункции sinθ
29
Для проверки равномерности распределения точек по сфере проводится разбиение сферы на области равной площади, совокупность которых полностью покрывает сферу. С помощью вершин вписанного в единичную сферу икосаэдра, соединенных геодезическими линиями, она может быть покрыта 20 одинаковыми равносторонними криволинейными треугольниками. Для исследования статистических свойств получаемых случайных распределений икосаэдр случайным образом поворачивается относительно сферы и при каждом новом его положении для одного и того же распределения определяется число точек, попавших в каждый криволинейный треугольник. Так получается представительная выборка попаданий точек на сфере в криволинейные треугольники, достаточная для анализа статистических характеристик распределения точек на сфере.
Анализ 1-го алгоритма распределения показывает наличие треугольников с большим числом точек, которые соответствуют областям вблизи полюсов сферы (см. рис. 6, а). При использовании 2-го алгоритма получается, что все накладываемые на сферу треугольники примерно одинаково заполняются точками распределения. На рис. 7 показаны виды функций распределения вероятностей. Представленные результаты демонстрируют узкую локализацию при использовании 2-го алгоритма. Функция, соответствующая 1-му алгоритму, имеет длинный «хвост». Таким образом, 2-й алгоритм, используемый для размещения точек по сфере, действительно позволяет получить равномерное распределение направления смещений атомов в пространстве.
Для заданного направления модуль смещения выбирается по равномерному закону из интервала [0;A] , где A – амплитуда воз-
мущений положений атомов. Такое распределение в пространстве не будет соответствовать равномерному заполнению шара: наблюдается большая концентрация возмущенных положений атома около центра шара. Последнее соответствует физическому смыслу процесса теплового движения атома вблизи равновесного положения.
30