

5605
.pdfПогрешность физико-химических методов составляет 2 – 5 %, что выше погрешности классических методов, но зато они позволяют определить микроколичества веществ (и часто в небольшой навеске).
1.3. Основные приёмы измерений
Измеряемые аналитические сигналы регистрируют тем или иным способом и получают регистрограммы, фотографии, наборы чисел и т.п. Для расчёта содержания исследуемого компонента обычно используют два основных методических приёма: метод прямых и косвенных измерений. К методу косвенных измерений относится титрование с инструментальным способом фиксирования точки эквивалентности.
При прямых измерениях используют зависимость аналитического сигнала от природы анализируемого вещества и его количества (концентрации). Часто эта зависимость выражается простым линейным соотношением y = ac, где a – константа, c – концентрация. Наибольшее распространение получили методы
сиспользованием стандартных веществ:
-метод градуировочного графика;
-метод молярного свойства;
-метод сравнения (стандарта);
-метод добавок.
Метод градуировочного (калибровочного) графика. В этом методе измеряется интенсивность аналитического сигнала (у) у нескольких стандартных образцов или стандартных растворов и строится градуировочный график обычно в координатах y = f(c), где с – концентрация определяемого компонента в стандартном растворе. Затем в тех же условиях измеряется интенсивность аналитического сигнала у анализируемой пробы ух и по градуировочному графику находится концентрация сх анализируемого вещества (рисунок 1.3.1).
Интервал концентрации на градуировочном графике должен охватывать предполагаемую область анализируемых концентраций, а состав стандартного образца или раствора должен быть близок к составу анализируемого. Например, при потенциометрическом определении нитрат-ионов в образце в качестве стандартного раствора может быть NaNO3 или KNO3.
11
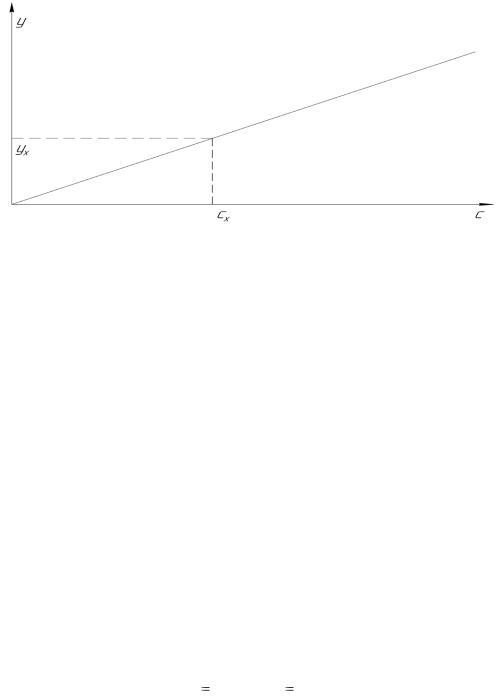
Рисунок 1.3.1 – Определение концентрации сх по градуировочному графику
Метод молярного свойства. Здесь также измеряется интенсивность аналитического сигнала у нескольких стандартных растворов и рассчитывается молярное свойство А, т.е. интенсивность аналитического сигнала, пропорциональная 1 моль вещества: А = у/с. Например, молярный коэффициент поглощения в спектрофотометрии. Затем в тех же условиях измеряется интенсивность сигнала у анализируемой пробы и по соотношению с = у/А рассчитывается концентрация анализируемого компонента. Метод предполагает строгое соблюдение соотношения у = Ас, по крайней мере, в области анализируемых концентраций.
Метод сравнения (метод стандарта). Используется в тех случаях, когда линия зависимости: состав – свойство (у-с) имеет прямолинейный характер и проходит через начало осей координат. На приборе измеряют интенсивность аналитических сигналов стандартного (уст) и исследуемого (ух) растворов. При этом отношение концентрации стандартного (сст) и анализируемого (сх) растворов равно отношению интенсивности аналитических сигналов:
ccт |
|
уст |
; сх |
сст |
у х |
. |
cx |
|
у х |
уст |
|||
|
|
|
|
Метод сравнения обладает меньшей точностью по сравнению с методом градуировочного графика, потому что в методе сравнения используется только одно измерение стандартного раствора.
Метод добавок. В этом методе сначала измеряется интенсивность аналитического сигнала пробы, затем в пробу вводится известный объём стандартного раствора до достижения концентрации сст и снова измеряется интенсивность сигнала. Если ух – интенсивность аналитического сигнала пробы,
12
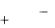
а у(х+ст.) – интенсивность сигнала после добавки стандартного раствора, то, очевидно,
Ух = Асх;
у(х+ст.) = А(сх + сст.);
сх = сст |
|
у х |
|
. |
|
|
|
||
|
у |
(х ст.) |
у х |
|
|
|
|
|
|
Метод также предполагает строгое соблюдение уравнения у = Ас и нередко выполняется графически.
Косвенные измерения – это методы титрования. В этих методах в ходе титрования измеряется интенсивность аналитического сигнала «у» и строится кривая титрования в координатах y – v, где v – объём добавленного титранта, мл. Точка эквивалентности находится на кривой титрования. Виды кривых титрования весьма многообразны, так как интенсивность аналитического сигнала может быть связана с концентрацией определяемого вещества, титранта или продукта реакции.
1.4. Подготовка аналитической пробы к измерительному анализу
Проведение измерительного анализа в лаборатории обычно складывается из нескольких этапов. Лабораторная проба, которая поступила в лабораторию, хотя
иявляется представительной пробой неоднородного образца часто требует некоторой дополнительной подготовки к анализу. Это связано главным образом с дополнительным измельчением и перемешиванием пробы. Из подготовленной лабораторной пробы отбирают аналитическую пробу, необходимую для единичного определения и взвешивают на аналитических весах. Масса аналитической пробы зависит от содержания определяемого компонента в пробе
ичувствительности применяемой методики определения.
Некоторые объекты не требуют никакой дополнительной обработки перед измерением аналитического сигнала. Например, измерение показателя преломления жидкости или раствора с помощью некоторых радиометрических методов, а также анализ изделий из металлов и сплавов методом эмиссионной спектроскопии. Но в большинстве случаев требуется подготовка пробы к анализу.
Так, при определении компонентов из твёрдых образцов их необходимо перевести в раствор. При этом под действием воды, кислот, оснований или
13
органических растворителей растворяется вся проба или происходит извлечение только определяемого компонента. Например, извлечение хлорида натрия из образца колбасы проводится путём встряхивания измельчённой пробы в течение нескольких минут, затем раствор отфильтровывают. Для некоторых объектов исследования требуются более сложные приёмы извлечения определяемых компонентов. Например, металлы и неметаллы в пищевых продуктах могут одновременно находиться в виде неорганических соединений, органических комплексов, металлорганических соединений и в других формах, в то время как аналитический сигнал при их определении многими физико-химическими методами (в частности, электрохимическим) обычно даёт только одна аналитическая форма. В этом случае требуется разложение пробы с целью перевода всех металлов и неметаллов в одинаковое ионогенное состояние.
Если в пробе содержится несколько подлежащих определению компонентов с близкими аналитическими сигналами или определению одного из них мешают другие, необходима операция разделения.
Кроме того, небольшое количество определяемого вещества в объекте часто требует его концентрирования. Таким образом, в подавляющем большинстве методов исследования перед определением необходима операция подготовки пробы, преследующая цели выделения, разделения и концентрирования вещества. Конечно, не в каждой методике реализуются все эти операции.
Решение вопроса оптимизации анализа в целом невозможно без оптимизации процесса подготовки проб, который часто является самым трудоёмким и лимитирующим скорость всего процесса исследования образца.
Общепринятыми методами выделения, разделения и концентрирования веществ являются минерализация, испарение, перегонка (дистилляция), экстракция, осаждение, ионный обмен, хроматография, электрофорез и электрохимическое концентрирование.
Так, для пищевых продуктов, основой которых являются белки или клетчатка, с целью выделения и концентрирования, например, азота или ионов металлов и неметаллов необходимо проводить минерализацию путём сухого или мокрого озоления.
Сухое озоление (сожжение) органических веществ проводят под действием кислорода воздуха или кислорода из баллона. Большинство пищевых продуктов сгорает при температуре 550 – 600ºС. Преимуществом метода сухого озоления является простота аппаратуры (термопечи и тигли). Недостатком – возможность потерь легколетучих компонентов (Hg, As и др.) и длительность процесса.
14
Получило широкое распространение сухое сожжение с озоляющими добавками. Это могут быть окислители, вещества, препятствующие улетучиванию. Так, например, белковые объекты предварительно обрабатывают азотной кислотой, затем озоляют. Объекты, основой которых является клетчатка (зерно, бобы, шоколад, кофе), вначале озоляют без кислоты, затем с добавлением азотной кислоты. Сухую минерализацию применяют, например, при определении общего фосфора в тканях, олова, свинца и железа в консервах.
Мокрое озоление. Мокрое озоление предполагает окисление органических веществ кислотами-окислителями и их смесями при повышенной температуре. Различают разложение в открытых сосудах (эффективность разложения зависит от температуры улетучивания кислот и их окислительной способности), а также в закрытых сосудах. В последних эффективность разложения повышается, так как с увеличением давления повышается температура кипения кислот и скорости реакций окисления, разложения соответственно растут. Для разложения пищевых продуктов чаще всего используют концентрированную азотную, смеси азотной и серной кислот, смеси HNO3 и H2O2, HNO3 и HClO4 в различных соотношениях. Соляная кислота не является окислителем и не разрушает большинство органических веществ, но растворяет органические вещества основного характера: амины, алкалоиды; разлагает металлорганические соединения Cu, Fe, Zn, Sn, As. Эти ионы выделяют из биологических жидкостей и пищевых продуктов экстракцией HCl. Такой приём используют, например, при определении кислотно-растворимого фосфора в мясе.
Преимуществом метода мокрого озоления перед сухим является большая скорость процесса, снижение потерь от улетучивания, особенно в закрытом варианте. Общим недостатком методов мокрого сожжения является использование токсичных концентрированных реагентов. В связи с длительностью процессов разложения органических веществ методами сухой и мокрой минерализации в последние годы стали использовать воздействие различных физических полей. Так, при анализе пищевых продуктов всё шире применяются возбуждённый кислород (низкотемпературная кислородная плазма), микроволновые поля, ультрафиолетовый свет, ультразвуковые воздействия и др.
Из методов испарения для концентрирования чаще применяют выпаривание. Пределы применения методов испарения обусловливаются устойчивостью определяемого вещества к температурным воздействиям, его летучестью,
15

окисляемостью. Путём испарения часто концентрируют водные и другие растворы, например, вина, водки, соки, безалкогольные напитки. Предварительно, в случае необходимости, вещество переводят в нелетучую форму. Если вещество неустойчиво при повышенных температурах, упаривание проводят в вакууме, что позволяет снизить температуру.
Аналитическая перегонка, или дистилляция, основана на испарении летучих компонентов, что позволяет провести их отделение и в случае необходимости, разделение или количественное определение. В связи с этим перегонка получила широкое развитие как метод выделения, определения и разделения летучих веществ (эфиров, спиртов и т.д.). Так, используя зависимость температуры кипения от состава смеси, определяют содержание этанола в смесях с водой.
Температуры кипения водно-спиртовых смесей различной концентрации: |
|
|||||
Этанол, в % |
100 |
80 |
62 |
41 |
20 |
0 |
t кип. ºС |
78,3 |
78,8 |
80,4 |
82,0 |
80 |
100 |
Особенно широко перегонку используют для выделения из материалов таких летучих компонентов, как эфирные масла. Аналитическая перегонка применяется для определения азота в органических соединениях методом Къельдаля и др.
Одним из наиболее часто применяемых методов является экстракция, которая используется в гетерогенной системе (твёрдая фаза-жидкость или жидкость-жидкость).
Если система состоит из двух несмешивающихся жидкостей, например, водный раствор – хлороформ, то избирательное извлечение веществ из одной жидкой фазы (чаще водной) в другую (органический растворитель) происходит в соответствии с законом распределения Нернста, который описывается уравнением
K D |
A орг. |
, |
|
A водн. |
|||
|
|
где КD – константа распределения; [A] орг. – концентрация вещества А в органической фазе; [A] водн. – концентрация вещества А в водной фазе, существует и иная форма записи этого уравнения:
D |
c A(орг.) |
, |
|
c A(водн.) |
|||
|
|
здесь D – коэффициент распределения с учётом всех форм существования вещества А в органической и водной фазе.
16
Эффективность разделения веществ с помощью экстракции оценивают по фактору разделения FА,В, который равен отношению коэффициентов распределения разделённых веществ А и В.
FA,B DA ,
DB
чем больше F, тем больше вещества А перейдёт в органическую фазу, а в водной будет больше содержание вещества В.
Концентрирование вещества происходит при извлечении вещества из большого объёма водной фазы в меньший – органической. Возможна и реэкстракция. Жидкостную экстракцию часто применяют для последующего фотометрического количественного определения извлечённого вещества.
Примером экстракции из твёрдой фазы в жидкую является извлечение веществ из растительных и животных тканей. При этом одновременно с экстракцией свежих тканей производят их гомогенизацию – размалывание ткани вместе с экстрагентом до полного разрушения клеток.
С целью концентрирования используют избирательную адсорбцию, особенно на ионообменных смолах (ионитах). Подобрав соответствующий ионит, можно избирательно адсорбировать необходимые ионы или органические основания или кислоты. После насыщения ионообменной колонки адсорбированное вещество смывают подходящим растворителем, получая более концентрированный раствор.
В электрохимическом концентрировании используют способность вещества подвергаться электрохимическому окислению или восстановлению. Например, в методе вольтамперометрии для концентрирования и повышения чувствительности метода применяют амальгамный электрод. При наложении на него отрицательного определённого потенциала концентрируемые катионы, восстанавливаясь, накапливаются в поверхностном слое амальгамного электрода. Изменив знак потенциала электрода, определяют присутствие и количество катионов.
Для разделения веществ часто применяют метод осаждения, используя при этом различие в растворимости определяемого и мешающих соединений. С помощью селективного осадителя можно перевести в осадок определяемое вещество, а мешающие компоненты останутся в растворе или наоборот. Например, осаждая белки трихлоруксусной кислотой, а углеводы солями тяжёлых металлов, выделяют молочную кислоту из мышечной ткани.
Одним из современных методов разделения и концентрирования близких по составу веществ является хроматография, которая основывается на использовании явлений сорбции в динамических условиях. Общий принцип всех многообразных
17
видов хроматографии состоит в том, что разделение веществ происходит вследствие неодинакового распределения их между подвижной и неподвижной фазами (подробнее см. в разделе «Хроматографический анализ»).
Многообразие методов выделения, разделения и концентрирования позволяет выбрать наиболее подходящий, а при необходимости сочетать некоторые из них с тем, чтобы сократить время для подготовки аналитической пробы к измерению аналитического сигнала соответствующим методом анализа.
В качестве примера приведём способы подготовки проб пищевых продуктов к анализу на содержание солей токсичных металлов методом инверсионной вольтамперометрии. Подготовка пробы в данном случае зависит от структуры матрицы. Так, в белковых объектах (молоко, рыба, мясо, яйца) матрицу разрушают путём обработки пробы азотной кислотой с последующим озолением. Объекты, основой которых является клетчатка (зерно, бобы, крупа, кофе и т.д.), измельчают и разрушение матрицы проводят сухим озолением до обугливания без кислоты, затем с добавлением азотной кислоты.
Продукты на основе углеводов, сахаров (сахар, карамель) измельчают в пудру и растворяют в соляной кислоте. Овощи и фрукты пропускают через соковыжималку и отфильтровывают. Винно-водочные изделия и безалкогольные напитки при необходимости концентрируют путём упаривания. Пищевые добавки: уксусную эссенцию, лимонную кислоту, поваренную соль – анализируют без пробоподготовки (навеску растворяют в бидистиллированной воде).
Глава 2. Спектральные и другие оптические методы анализа
Спектральные и другие оптические методы анализа основаны на использовании различных явлений и эффектов, возникающих при взаимодействии вещества и электромагнитного излучения. Рассмотрим метод
абсорбционной спектроскопии.
Абсорбционная спектроскопия делится на молекулярную и атомную. К молекулярной абсорбционной спектроскопии относится спектрофотометрия (фотометрия), основанная на поглощении веществом электромагнитного излучения в видимой, ультрафиолетовой и инфракрасной части спектра, т.е. оптического диапазона (от 100 до 100 000 нм).
2.1. Основной закон светопоглощения
18
Молекула, поглощая квант света, переходит в более высокое энергетическое состояние. Обычно это бывает переход с основного, невозбуждённого уровня на один из более высоких, чаще всего на первый возбуждённый уровень. Вследствие поглощения излучения при прохождении его через слой вещества интенсивность излучения уменьшается и тем более, чем выше концентрация светопоглощающего вещества и толщина слоя. Эту зависимость отражает закон Бугера – Ламберта – Бера (основной закон светопоглощения).
Поток монохроматического излучения с интенсивностью Jº проходя через слой раствора с концентрацией поглощающих частиц «с» и толщиной l ослабляется (рисунок 2.1.1). Поглощение излучения происходит пропорционально толщине слоя и концентрации поглощающих частиц.
Рисунок 2.1.1 – Прохождение света через раствор
Уменьшение интенсивности света, прошедшего через раствор, характеризуется пропусканием Т и оптической плотностью А (по-старому Д).
J J 10 |
|
kcl |
или |
lg |
J |
|
kcl ; |
||
|
|
|
|
J |
|||||
|
|
|
|
|
|
|
|
||
lg |
J |
|
A; A |
kcl ; |
|
||||
|
J |
|
|||||||
|
|
|
|
|
|
|
|||
T |
|
J |
; |
А |
lgT . |
|
|||
|
|
|
|||||||
|
|
J |
|
|
|
|
|
||
Важной характеристикой является коэффициент поглощения «k», который зависит от природы вещества и растворителя, длины волны падающего света и температуры.
Физический смысл коэффициента поглощения становится ясным, если принять коэффициент поглощения раствора и толщину поглощающего слоя за единицу. Тогда k = A. При этих условиях «k» представляет собой поглощение раствора и является специфической характеристикой вещества. Если концентрация раствора выражается в моль/дм3 и толщина слоя в см, то «k» называется молярным коэффициентом поглощения и обозначается буквой ε. Тогда
19
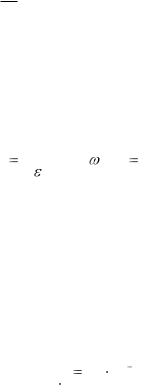
А = εсl . |
(2.1) |
Если концентрация вещества выражена в массо-объёмных долях (ω/), то «k» называют удельным показателем поглощения и обозначают E1%1см . Обе величины
связаны между собой ε = E1% М .
1см 10
Значения молярного и удельного коэффициентов поглощения как специфические характеристики вещества используют для целей идентификации. Эти величины используют также для определения чувствительности спектрофотометрических определений и для количественного определения по уравнению связи:
Cmin |
Amin |
; |
/ |
Amin |
, |
l |
min |
1% |
|||
|
|
|
|
E1см |
|
где сmin – минимальная концентрация, поддающаяся спектрофотометрическому определению моль/дм3, Аmin – минимальное значение оптической плотности, регистрируемое прибором (обычно 0,01). Например, при фотометрическом определении железа (III) с помощью сульфосалициловой кислоты в кислой среде при λmax = 510 нм, при толщине слоя 1 см (ε = 1,8 ∙ 103) минимально определяемая концентрация Fe3+ равна:
Сmin = |
0,01 |
5,5 10 |
6 моль/дм3 . |
||
|
|
||||
1800 1 |
|||||
|
|
|
|||
Чем больше молярный или удельный коэффициент поглощения, тем чувствительнее методика определения. Молярные коэффициенты некоторых соединений достигают 100 000 – 120 000.
Оптическая плотность раствора, содержащего несколько поглощающих веществ, обладает свойством аддитивности, которое иногда называют законом аддитивности светопоглощения. В соответствии с этим законом поглощение света каким-либо веществом не зависит от присутствия в растворе других веществ. При наличии в растворе нескольких поглощающих веществ каждое из них будет давать свой аддитивный вклад в экспериментально определяемую оптическую плотность А:
А= А1 + А2 + … Аn или А = l(ε1c1 + ε2c2 + … εncn ).
2.2.Ограничения и условия применимости закона Бугера – Ламберта – Бера
Всоответствии с уравнением (2.1) зависимость оптической плотности от концентрации выражается прямой линией, выходящей из начала координат
20