

2.3. Электрохимическая коррозия
2.3.1. Причины возникновения коррозии
Электрохимическая коррозия металла может проявиться во всех тех случаях, когда имеет место граница раздела фаз металл - электролит. Факт появления коррозии не зависит от природы электролита, будь это сверхчистая вода или расплавленная соль. Не имеет существенного значения и количество электролита - в предельном случае это может быть пленка влаги толщиной в несколько десятков миллимикрон. Единственным условием является возможность совмещения на металлической поверхности анодной реакции ионизации металла и катодной реакции восстановления тех или иных ионов или молекул. Такая возможность реализуется, если равновесный потенциал анодной реакции окажется отрицательнее равновесного потенциала хотя бы одной катодной реакции. Тогда установившийся стационарный потенциал займет промежуточное положение.
Это условие обязано соблюдаться независимо от разновидности коррозии - от атмосферной до коррозии под действием блуждающих токов. В каждом из случаев мы будем иметь дело с частным проявлением неравновесных электродных процессов и можем объяснить механизм протекания реакций с позиции закономерностей электрохимической кинетики. Поэтому саму теорию коррозии удобно называть кинетической или теорией Фрумкина - Вагнера, по имени ученых, разработавших основные ее принципы.
Теория
процесса.
Важно уяснить, что кинетическая теория
коррозии справедлива применительно
даже к идеально чистому металлу.
Действительно, при погружении чистого
металла в водный раствор его соли
устанавливается равновесие Ме ↔ Меn+
+
nе,
отвечающее равновесному потенциалу .
Это означает, что скорости ионизации
![]() и разряда
и разряда ![]() равны и составляют ток обмена
равны и составляют ток обмена![]() . Равновесное состояние свидетельствует
об отсутствии коррозии, что возможно
только в том случае, если предполагаемый
в данных условиях катодный процесс
(например, восстановление водородного
иона) имеет равновесный потенциал
. Равновесное состояние свидетельствует
об отсутствии коррозии, что возможно
только в том случае, если предполагаемый
в данных условиях катодный процесс
(например, восстановление водородного
иона) имеет равновесный потенциал
![]() отрицательнее потенциала ионизации
металла.
отрицательнее потенциала ионизации
металла.
Если,
наоборот,
![]() ,
то на границе раздела фаз металл -
раствор устанавливается равновесие
,
то на границе раздела фаз металл -
раствор устанавливается равновесие
![]() +
+![]() =
=![]() +
+![]() ,
,
соответствующее уже двум различным реакциям:
Me ↔ Mn+ + ne,
nH+ + ne ↔ n/2H2.
При этом, исходя из неравенства, имеем
> и < .
В результате будет происходить преимущественно ионизация, или разрушение, металла при стационарном потенциале, который соответствует неравенству
![]() .
.
Если ток обмена металла по абсолютной величине превышает ток обмена водорода (поляризуемость катодного процесса больше из-за большего перенапряжения выделения водорода), то стационарный потенциал приблизится к равновесному потенциалу металла.
Этот
вариант показан схематично на рис. 25,а
и
представлен поляризационными кривыми
рис. 25, б.
На схеме сумма векторов тока ионизации
металла и водорода равна сумме векторов
тока их восстановления. Следовательно,
стационарный потенциал установился. В
то же самое время абсолютная величина
скорости каждой из электродных реакций
с участием металла заметно превышает
скорости реакций с участием водорода.
Это связано со значительным различием
токов обмена
и
![]() ,
которые не сильно отличаются от
соответствующих токов ионизации и
разряда. Судя по рис. 25, б,
коррозия металла протекает со
скоростью ia,
а выделение водорода происходит с
равной ей скоростью ik.
,
которые не сильно отличаются от
соответствующих токов ионизации и
разряда. Судя по рис. 25, б,
коррозия металла протекает со
скоростью ia,
а выделение водорода происходит с
равной ей скоростью ik.
Аналогичным образом можно показать, что при токе обмена водорода, превышающем ток обмена металла, стационарный потенциал приближается к равновесному потенциалу водорода. В этом случае перенапряжение реакции ионизации металла будет выше перенапряжения выделения водорода.
Анодный процесс при коррозии всегда заключается в ионизации металла. В катодном процессе могут участвовать разнообразные ионы или молекулы, являясь по отношению к металлу окислителями.
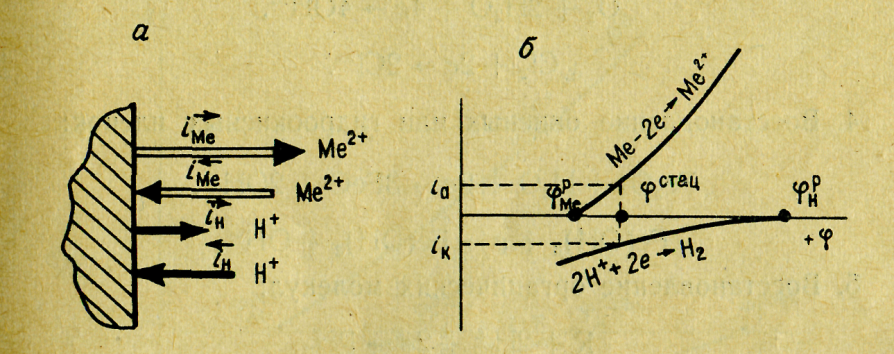
Рис. 25. Схема установления стационарного потенциала коррозии при токе обмена металла выше тока обмена водорода (а) и поляризационная диаграмма сопряженных электродных реакций (б)
Происходят следующие основные типы катодных реакций:
Восстановление катиона:
H+ + e →1/2H2,
Ag+ + e →Ag.
2. Восстановление аниона:
S2O82- + 2e → 2SO42-,
MnO-4 + e → MnO42-.
3. Восстановление молекул растворенного в электролите газа:
О2 + 2Н2О + 4e → 4OH-,
Cl2 + 2e → 2Cl-.
Восстановление оксидных или гидроксидных пленок
Fе3О4 + Н2О + 2е → 3FеО + 20Н-,
Fе(ОН)3 + е → Fе(ОН)2 + ОН-.
Восстановление органических молекул:
R + 2Н+ + 2е → RH2,
RО + 4Н+ + 4е → RН2 + Н2О.
В электролите, как правило, присутствуют два или несколько окислителей. Так, в водном растворе наряду с водородными ионами всегда содержится некоторое количество растворенного кислорода и нередко органические примеси. Их совместное влияние на скорость анодного процесса будет зависеть от взаимного расположения катодных поляризационных кривых по отношению к анодной кривой. Рассмотрим случай с двумя окислителями.
На рис. 26 даны поляризационные кривые КI и КII, отвечающие двум катодным реакциям, протекающим с близким перенапряжением.
Согласно принципу аддитивности поляризационных кривых можно построить суммарную катодную кривую КI+II. В присутствии только окислителя I скорость коррозии была бы равна iI при стационарном потенциале φIстац, в присутствии только окислителя II - скорость равна iII при потенциале φIIстац .
Но
если равновесный потенциал φIстац
менее активного окислителя положительнее
стационарного потенциала φIIстац
более активного (см. рис. 26), в коррозионном
процессе принимают участие оба окислителя.
Скорость коррозии при этом оказывается
выше, чем в более активном окислителе.
Действительно, из рисунка видно, что
iI+II
>iI
>iII
. В то же время доля участия каждого из
окислителей снижается, так как стационарный
потенциал
![]() сместился вправо, в сторону меньшего
катодного тока. Иными словами, при
совместном участии в коррозионном
процессе двух катодных реакций скорость
каждой из реакций становится ниже, чем
при раздельном участии: ΔiI
<
iI
и ΔiII
< iII.
сместился вправо, в сторону меньшего
катодного тока. Иными словами, при
совместном участии в коррозионном
процессе двух катодных реакций скорость
каждой из реакций становится ниже, чем
при раздельном участии: ΔiI
<
iI
и ΔiII
< iII.
Технически чистые металлы всегда загрязнены примесями, а сплавы содержат еще и легирующие добавки; поверхность технических металлов характеризуется структурной и термодинамической неоднородностью. Поэтому коррозия в реальных условиях - это коррозия многокомпонентного металла с неравновесным состоянием поверхности.
Каждому из металлических включений, каждому из термодинамически неоднородных участков соответствуют свои равновесные потенциалы катодных и анодных реакций и своя поляризуемость. Результирующая скорость коррозии, как и сама возможность коррозии основного металла или металла-примеси, определяется соотношением всех поляризационных кривых.
В качестве характерного примера рассмотрим случай коррозии металла МеI, имеющего включения металла МеII. Поляризационные анодные кривые, как видно из рис. 26, заметно отстоят одна от другой; металл МеII значительно электроположительнее металла МeI.
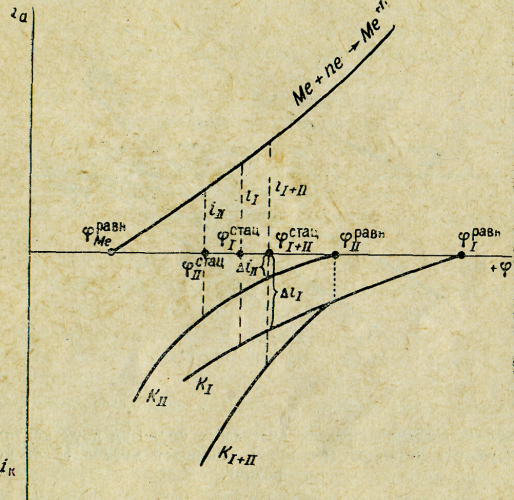
Рис. 26. Поляризационная диаграмма коррозии металла в присутствии двух окислителей (оба окислителя участвуют в коррозионном процессе)
Для простоты допустим, что сопряженной катодной реакцией для каждого металла может быть только восстановление водорода.
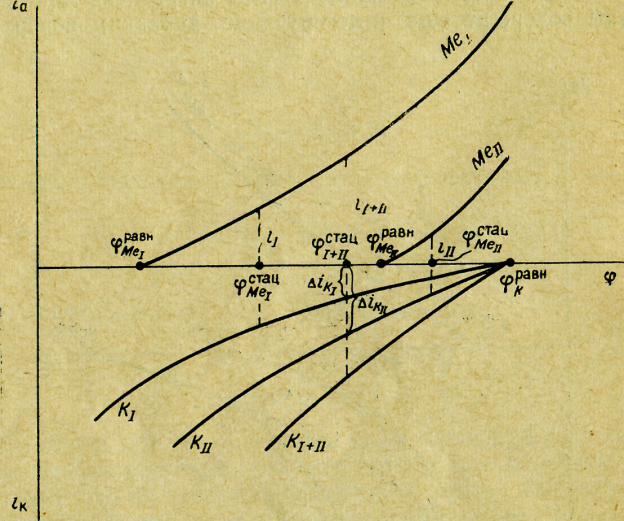
Рис. 27. Поляризационная диаграмма коррозии двух контактирующих металлов (металл I защищает металл II от коррозии)
В
случае полной электрической изоляции
каждый металл корродировал бы с
определенной и взаимно независимой
скоростью iI
и iII.
Но металлы находятся в контакте. Поэтому
катодная поляризационная кривая
равна суммарной КI+II
.При
этом оказывается, что скорость коррозии
возросла до iI+II,
но корродирует только металл МеI
, потому что стационарный потенциал
,
отрицательнее равновесного потенциала
![]() .
Что касается водорода, то он выделяется
как на металле MeI,
так и на металле МеII,
но с неодинаковой скоростью. Скорость
катодной реакции
.
Что касается водорода, то он выделяется
как на металле MeI,
так и на металле МеII,
но с неодинаковой скоростью. Скорость
катодной реакции ![]() гораздо
ниже, чем скорость реакции
гораздо
ниже, чем скорость реакции
![]() .
.
В результате коррозии по схеме рис. 27 в цепи МеI - MeII протекает, несмотря на контакт металлов, электрический ток. Если переходное сопротивление в месте контакта ощутимо, то каждый металл будет обладать своим стационарным потенциалом, отличающимся от . Скорость коррозии при этом будет снижаться за счет омических потерь на границе МeI /МеII. Такой же эффект будет оказывать понижение электропроводности электролита (коррозионной среды).
Ток, протекающий в системе металл - электролит -металл, называется локальным, а сама система представляет собой своеобразный гальванический элемент, включенный накоротко. Теория, объясняющая механизм коррозии работой многочисленных макро- и микроэлементов, как известно, возникла в 30-е годы прошлого столетия и связана с именем де ля Рива. В дальнейшем она успешно развивалась благодаря усилиям многих крупнейших ученых, в том числе Акимова и Эванса. Теория локальных элементов подкупает убедительностью и доступностью понимания. Так, качественные закономерности коррозии удобно изучать на моделях гальванических элементов. Получаемые результаты весьма наглядны в виде коррозионных диаграмм потенциал-ток (диаграммы Эванса).
В то же время теория де ля Рива – Эванса - Акимова, как основанная на принципе пространственного разделения катодных и анодных участков, не вполне отвечает современным представлениям о механизме электродных реакций. Количественные расчеты в рамках этой теории крайне затруднены, так как далеко не просто определить суммарные поверхности всех катодных и всех анодных участков металла в отдельности. Нередко некоторые участки являются катодными в системе одной пары и анодными в системе другой.
Согласно теории локальных элементов окислители, обеспечивающие протекание катодной реакции, называются деполяризаторами, а сам катодный процесс деполяризацией.
И все же теория локальных элементов как теория электрохимическая не должна противопоставляться кинетической теории. Скорее наоборот, эти теории эффективно дополняют одна другую при сохранении ведущего положения за кинетической теорией.