

В процессе развития апоптоза различают следующие стадии:

Сравнительная характеристика апоптоза и некроза
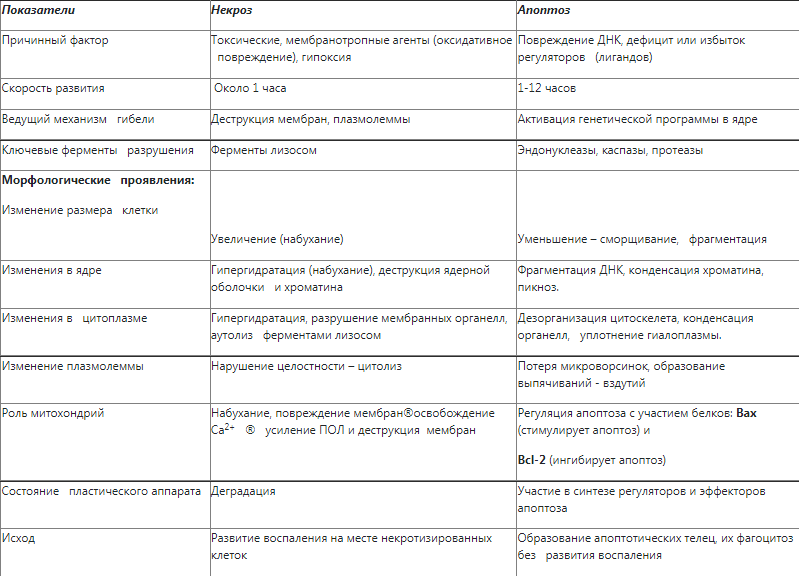
Внешний и внутренний механизмы развития апоптоза
Развитие апотоза может быть вызвано действием внеклеточных факторов или индуцировано изменением внутриклеточного гомеостаза, что определяет наличие 2 путей индукции апоптоза:
- внешнего – через Fas-систему плазмолеммы (через рецепторы смерти);
- внутреннего – через Bax-систему митохондрий.
Внутренний (митохондриальный) механизм развития апоптоза
Данный механизм считается ведущим и связан с увеличением проницаемости митохондриальных мембран, ведущим к освобождению в цитоплазму клетки митохондриальных белков. Освобождение данных белков регулируется целым семейством про- и антиапоптогенов, многие из которых относятся к семейству Bcl. Факторы роста и субстраты стимулируют продукцию антиапоптогенов (Bcl-2, Bcl-x, и Mcl-1). Эти белки входят в состав цитоплазмы и митохондриальных мембран, контролируют проницаемость митохондриальных мембран и предотвращают освобождение митохондриальных белков, вызывающих гибель клетки. При отсутствии необходимых факторов роста и субстратов происходит активация сенсоров повреждения и смерти. Эти сенсоры также относятся к семейству Bcl и включают так называемые белки Bim, Bid и Bad. Эти белки в свою очередь активируют два критических проапоптогенных эффектора - Bax и Bak. Данные проапоптогены являются олигомерами и встроены в митохондриальные мембраны, формируя каналы (поры), обеспечивающие выход белков из внутренней митохондриальной мембраны в цитозоль.
Активация Bax-Bak ведет к выходу в цитозоль цитохрома с (вовлеченного в цепь переноса электронов) и др, что включает каспазный каскад. Освобожденный цитохром с связывается с белком Apaf-1 (apoptosis-activating factor-1),и формирует гексамерную структуру, названную апоптосомой apoptosome. Данный комплекс связывается с каспазой-9, критерием инициатором митохондриального пути апоптоза.
Внешний (опосредованный через рецепторы смерти) механизм развития апоптоза
Данный путь апоптоза связан с наличием на плазмолемме рецепторов смерти – это рецепторы к фактору некроза опухолей (ФНО). Данное семейство рецепторов содержит домен смерти. Наиболее изученным среди них является 1 тип рецепторов к ФНО (TNFR1), называемый Fas (CD95). Механизм апоптоза связан с взаимодействием лиганд с рецепторов (лиганд Fas (FasL)). Fas При связи FasL с Fas, три и более молекул Fas объединяются вместе и их цитоплазматическая часть (домен смерти) присоединяется к адаптерному белку FADD (Fas-associated death domain). FADD в свою очередь связывается с каспазой-8 и каспазой-10. Активация капсаз обеспечивает реализацию апоптоза.
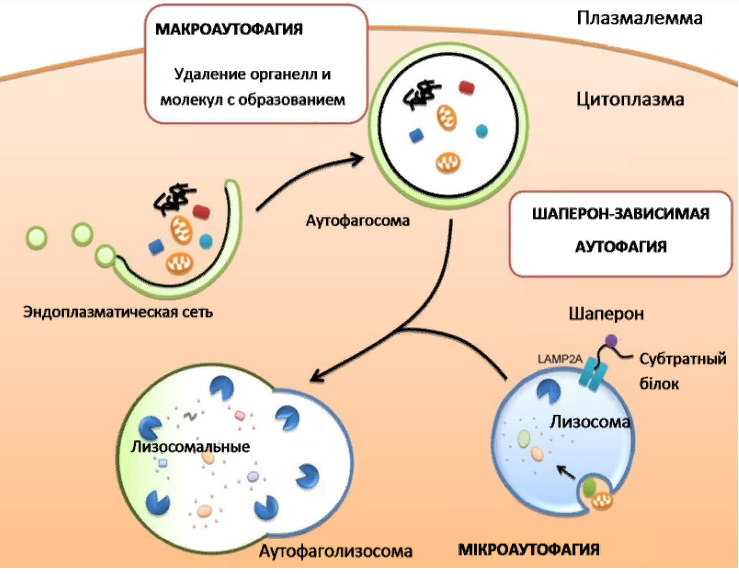
Аутофагия
Помимо некроза и апоптоза в последние десятилетия был открыт еще один вид клеточной гибели – аутофагия.
Аутофагия (или каспаза-независимый апоптоз) — один з способов избавления клеток от ненужных молекул и органелл, а также путь ликвидации из организма ненужных клеток. Аутофагия не всегда ведет к гибели клетки. Выделяют:
- Макроаутофагия – фрагмент цитоплазмы с органеллами охватывается двойными мембранами с образованием аутофагосом, которые далее сливаются с лизосомами, образуя аутофаголизосому, где происходит лизис старых органелл и молекул.
- Микроаутофагия – макромолекулы и фрагменты клеточных органелл напрямую захватываются лизосомами. Таким образом клетка может использовать собственные белки для катаболизма.ю например, при голодании.
- Шаперон-зависимая аутофагия – обеспечивает направленный транспорт в лизосомы частично денатурированных би неправильно конформированных белков Данный вид аутофагии активируется при стрессе. Участниками данного процесса являются цитоплазматические белки шапероны (из семейства белков теплового шока hsc-70) и белки LAMP-2 – рецепторы мембран лизосом к комплексу шаперон+аномалный белок, подлежащий деградации.
Аутофагия реализуется в норме и при реакции клетки на повреждение. Факторами, усиливающими аутофагию являются: дефицит питательных веществ (субстратов), наличие в цитоплазме поврежденных органелл, денатурованных белков и их агрегатов, клеточный стресс (при ишемии, действии механических и токсических факторов). При аутофагической гибели клеток происходит разрушение всех орагенлл и ядра. Остается только клеточный дебрис, который фагоцитируют макрофаги.
Физиологическое и патофизиологическое значение аутофагии:
аутофагия вовлечена в регуляцию развития органов в эмбриогенезе.
Принимает участие в физиологической и репаративной регенерации
Снижение аутофагии лежит в основе развития возраст-ассоциирвоанной патологи за сет накопления в клетках старих органелл и белков
Поддерживает функцию клеток в условиях дефицита субстратов (при голодании) путем утилизации собственых белков и органелл.
Ингибирует рост опухолей – за сет усиления катаболизма «неправильных» белков и деградации поврежденных органелл, ограничивая рост и пролиферацию опухолевых клеток.
Нарушение аутофагии – одна из причин развития врожденной и приобретенной патологи. Важную роль нарушение аутофагии играет при развитии миопатий и нейродегенеративных забоелваний, включая болезнь Альцгеймера, Хантингтона и Паркинсона. Болезнь Альцгеймера (сенильная деменция альцгеймеровского типа) — наиболее распространенная форма деменции, нейродегенеративное заболевание, впервые описанное в 1906 году немецким психиатром Алоисом Альцгеймером. Болезнь проявляется прогрессиреющим снижением памяті, агнозией, нарушением речевой функции и движений. В основе развития этой патологи лежит наруение протеасомной деградации белков и аутофагии. Микроскопически в зонах поражения мозга в отростках нейронов наблюдаетс накопление незрелых аутофагосом, что связано с нарушением их транспорта ис слияния с лизосомами. Следствеим этого является предотвращение разрушения старих и аномальних белков и органелл, накопление в цитоплазме аномальных белков и агрегатов в виде нейрофибриллярных клубков и бляшек. Аналогичные механизми лежат в основе розвития хореи Хаттингтона и болезни Паркинсона. Мутантные белки хантингтин и альфа-синуклеин — белки, накопление колторых вызывает развитие соотвественно болезни Гентингтона и Паркинсона — в норме удаляются в нейронах с помощью шаперон-зависимой аутофагии. При ее нарушении происходит накопление аномальных агрегатов в цитоплазме нейронов.