

Вопрос 103 Гемоглобины человека, структура. Транспорт кислорода и диоксида углерода. Гемоглобин плода и его физиологическое значение. Гемоглобинопатии.
Гемоглобины взрослого человека:
Гемоглобин составляет 90% от всех белков эритроцитов
Гемоглобин А – основной гемоглобин взрослого организма, составляет около 98%
от общего количества гемоглобина, тетрамер, состоит из 2 полипептидных цепей α и 2 β (2α2β).
Гемоглобин A2 находится в организме взрослого человека в меньшей
концентрации, на его долю приходится около 2% общего гемоглобина. Он состоит из 2 α- и 2 δ-цепей.
Гемоглобин А1с – гемоглобин А, модифицированный ковалентным
присоединением к нему глюкозы (так называемый гликозилированный гемоглобин).
Строение протомеров гемоглобина: протомеры состоят из 8 спиралей,
свёрнутых в плотную глобулярную структуру, содержащую внутреннее гидрофобное ядро и «карман» для связывания гема.
Четвертичная структура гемоглобина: 4 ППЦ образуют почти правильную
форму шара, где каждая α-цепь контактирует с двумя β-цепями. В области контакта между α- и β цепями находится много гидрофобных радикалов – формируется сильное соединение за счёт возникновения гидрофобных, ионных и водородных связей.
В результате образуются димеры α1β1 и α2β2.
Между этими димерами в тетрамерной молекуле гемоглобина возникают в
основном полярные (ионные и водородные) связи, поэтому при изменении pH среды в кислую или щелочную сторону в первую очередь разрушаются связи между димерами. Кроме того, димеры способны легко перемещаться относительно друг друга.
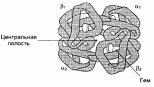
Т.к. поверхность протомеров неровная, ППЦ в центральной
области не могут плотно прилегать друг к другу, в результате в центре формируется «центральная полость», проходящая сквозь всю молекулу гемоглобина.
ТРАНСПОРТ КИСЛОРОДА И ДИОКСИДА УГЛЕРОДА
В крови СО2, образовавшийся в тканях, транспортируется в эритроциты.
В эритроцитах под действием фермента карбангидразы происходит увеличение
скорости образования Н2СO3. Слабая угольная кислота может диссоциировать:
СO2 + Н2O <=> Н2СO3 <=> Н+ + HCO3-
Равновесие реакции в эритроцитах, находящихся в капиллярах тканей, смещается
вправо, т.к. образующиеся в результате диссоциации H2CO3 протоны могут присоединяться к специфическим участкам молекулы гемоглобина: к радикалам Гис146 двух β-цепей, радикалам Гис122 и концевым α-аминогруппам двух α-цепей.
Все эти 6 участков при переходе гемоглобина от окси- к дезоксиформе
приобретают большее сродство к Н+ в результате локального изменения аминокислотного окружения вокруг этих участков (приближения к ним отрицательно заряженных карбоксильных групп аминокислот).
Присоединение 3 пар протонов к гемоглобину уменьшает его сродство к O2 и
усиливает транспорт O2 в ткани, нуждающиеся в нём
В капиллярах лёгких высокое парциальное давление О2 приводит к
оксигенированию гемоглобина и удалению 6 протонов.
Бикарбонаты с помощью белка 3 полосы обмениваются на Cl - и выходят
в плазму крови, образуется Н2СО3, который разрушается до СО2 + Н2О
Реакция СО2 + Н2О <=> Н2СО3 <=> Н++ НСО3- сдвигается влево и образующийся
СО2 выделяется в альвеолярное пространство и удаляется с выдыхаемым воздухом
Эффект Бора: увеличение освобождения O2гемоглобином в зависимости от
концентрации Н+ . В капиллярах лёгких высокое парциальное давление O2 приводит к
оксигенированию гемоглобина и удалению 6 протонов.
Реакция сдвигается влево и образующийся СO2 выделяется в альвеолярное
пространство и удаляется с выдыхаемым воздухом

Большая часть СO2 транспортируется кровью в виде бикарбоната НСO3-.
Присоединение СO2 к гемоглобину также снижает его сродство к O2.
2,3-Бифосфоглицерат (БФГ) – аллостерический регулятор сродства гемоглобина
к O2, вещество, синтезируемое в эритроцитах из промежуточного продукта окисления глюкозы 1,3-бифосфоглицерата.
БФГ, присоединяясь к гемоглобину, также может менять его сродство к O2.
Центральная полость тетрамера гемоглобина – место присоединения БФГ.
Размеры центральной полости могут меняться: отщепление O2 от оксигемоглобина
вызывает его конформационные изменения, которые способствуют образованию дополнительных ионных связей между димерами α1β1 и α2β2. В результате пространственная структура дезоксигемоглобина становится более жёсткой, напряжённой, а центральная полость расширяется.
Поверхность полости ограничена остатками аминокислот, в числе которых
имеются положительно заряженные радикалы Лиз82, Гис143 β-цепей и положительно заряженные α-аминогруппы N-концевого валина β-цепей.
В расширенную полость дезоксигемоглобина БФГ, имеющий сильный
отрицательный заряд, присоединяется с помощью ионных связей, образующихся с положительно заряженными функциональными группами двух β-цепей гемоглобина. Присоединение БФГ ещё сильнее стабилизирует жёсткую структуру дезоксигемоглобина и снижает сродство белка к O2
В лёгких высокое парциальное давление O2 приводит к оксигенированию
гемоглобина. Разрыв ионных связей между димерами α1β1 и α2β2 приводит к «расслаблению» белк. молекулы, уменьшению центральной полости и вытеснению БФГ.
Изменение концентрации БФГ как механизм адаптации организма к гипоксии.
Концентрация БФГ в эритроцитах людей величина постоянная. Но в период
адаптации к высокогорью, концентрация БФГ уже через 2 дня возрастает почти в 2 раза, что снижает сродство гемоглобина к O2 и увеличивает количество O2, транспортируемого в ткани. Такую же адаптацию наблюдают у больных с заболеваниями лёгких, при которых развивается общая гипоксия тканей.
ГЕМОГЛОБИНЫ ПЛОДА:
Эмбриональный гемоглобин синтезируется в эмбриональном желточном мешке
через несколько недель после оплодотворения. Представляет собой тетрамер 2α2ε. Через 2 нед после формирования печени плода в ней начинает синтезироваться гемоглобин F, который к 6 мес замещает эмбриональный гемоглобин.
Гемоглобин F- фетальный гемоглобин, синтезируется в печени и костном мозге
плода до периода его рождения. Имеет тетрамерную структуру, состоящую из 2 α- и 2 γ-цепей. После рождения ребёнка постепенно замещается на гемоглобин А, который начинает синтезироваться в клетках костного мозга уже на 8-м месяце развития плода.
В физиологических условиях его сродство к О2 выше, чем сродство к О2 у
гемоглобина А. Более высокое сродство к О2 гемоглобина создает оптимальные условия для транспорта кислорода из крови матери в кровь плода. Дезоксиемоглобин оксигенируется за счет гемоглобина А (находящегося по другую сторону трансплацентарного барьера в кровеносной системе плаценты).
Гемоглобин F слабее связывает БФГ, чем гемоглобин А, и, следовательно, обладает
более высоким сродством к O2. Физиологические особенности НbF связаны с особенностями его строения: вместо β-глобиновых цепей в НbА, он содержит две y-цепи
Связывание 2,3-БФГ с НbА происходит при участии положительно заряженных
радикалов аминокислот двух β-цепей, некоторые из которых отсутствуют в первичной структуре y-цепей. В среде, лишённой 2,3-БФГ, НbА и НbF проявляют одинаковое высокое сродство к O2.
ГЕМОГЛОБИНОПАТИИ
Гемоглобин А – тетрамер, состоящий из двух α- и двух β-цепей (2α2β)
В молекуле гемоглобина S (аномальный гемоглобин) мутантны две β-цепи, которых глутамат (высокополярная отрицательно заряженная АК) в положении 6 была заменена валином, содержащим гидрофобный радикал
В дезоксигемоглобине S имеется участок, комплементарный другому
участку таких же молекул, содержащему замененную АК. В результате молекулы дезоксигемоглобина начинают «слипаться», образуя удлиненные агрегаты, деформирующие эритроцит и приводящие к обр-ю эритроцитов в виде серпа
В оксигемоглобине S комплементарный участок недоступен для взаимодействия
из-за изменения конформации белка, поэтому молекулы оксигемоглобина S не слипаются.
Т.о. образованию агрегатов дезоксигемоглобина S способствуют условия,
повышающие конц. дезоксигемоглобина в клетках (физическая работа, гипоксия и др.)
Серповидные эритроциты плохо проходят через капилляры, часто закупоривают
сосуды и тем самым создают локальную гипоксию => повышается конц. дезоксигемоглобина S в эритроцитах, что увеличивает скорость образования агрегатов и вызывает еще большую деформацию эритроцитов (это может вызывать боль и некроз клеток в данной области)
1. Серповидно-клеточная анемия – гомозиготное рецессивное заболевание;
проявляется только в том случае, когда от обоих родителей наследуются 2 мутантных гена β-цепей глобина. Болезнь с рождения не проявляется до тех пор, пока значительное количество гемоглобина F (фетальный г., который после рождения обычно заменяется на HbA) не заместится на HbS. Возникают симптомы, характерные для анемии: головокружение и головные боли, учащенные сердцебиение, боли в конечностях и др.
Гетерозиготные индивидуумы, имеющие один нормальный ген HbA, а другой HbS
имеют лишь следовые кол-ва серповидных эритроцитов и нормальную продолжительность жизни.
Особенность: в Африке высока частота гена HbS, это связно с тем, что
гетерозиготные носители гена HbS менее чувствительны к малярии, чем люди с нормальным гемоглобином, т.к. их эритроциты имеют короткий срок жизни, поэтому возбудитель малярии не успевает закончить необходимую стадию развития (что и дает преимущество людям, гетерозиготным по HbS)
2. Талассемии – наследственные заболевания, обусловленные отсутствием или
снижением скорости синтеза α- или β-цепей гемоглобина. В результате несбалансированного образования глобиновых цепей образуются тетрамеры гемоглобина, состоящие из одинаковых протомеров, что приводит к нарушению транспорта кислорода к тканям. Нарушение эритропоэза и ускоренный гемолиз эритроцитов и клеток-предшественников при талассемиях приводит к анемии.
При β-талассемии не синтезируются β-цепи гемоглобина. Это вызывает
образование нестабильных тетрамеров, содержащих только α-цепи. При этом заболевании в костном мозге из-за преципитации нестабильных α-цепей усиливается разрушение эритробластов, а ускорение разрушения эритроцитов в циркулирующей крови приводит к внутрисосудистому гемолизу. Как известно, для образования фетального гемоглобина
β-цепи не требуются поэтому клинически β-талассемия не проявляется до рождения, после чего происходит переключение синтеза HbF на НBА.
В случае α-талассемии недостаток образования α-глобиновых цепей приводит к
нарушению образования HbF у плода. Избыточные γ-цепи образуют тетрамеры, называемые гемоглобином Барта. Этот гемоглобин при физиологических условиях имеет повышенное сродство к кислороду и не проявляет кооперативных взаимодействий между протомерами. В результате гемоглобин не обеспечивает развивающийся плод необходимым количеством кислорода, что приводит к тяжёлой гипоксии. При α-талассемии отмечают высокий процент внутриутробной гибели плода.
Выжившие новорождённые при переключении с γ- на β-ген синтезируют β-
тетрамеры или НBН, который, подобно гемоглобину Барта, имеет слишком высокое сродство к кислороду, менее стабилен, чем НBА и быстро разрушается. Это ведёт к развитию у больных тканевой гипоксии и к смерти вскоре после рождения.
3. Наследственный сфероцитоз - причиной является дефект белков цитоскелета
эритроцитов – спектрина или анкирина, которые обеспечивают поддержание двояковогнутой формы клетки и эластичности мембраны. Эритроциты приобретают шарообразную форму, что приводит к уменьшению площади их поверхности и снижению скорости газообмена.
Потеря эластичности клеточной мембраны приводит к повышению хрупкости и
травматичности клеток и, как следствие, к ускорению их разрушения в сосудистом русле и селезёнке. Заболевание сопровождается анемией и желтухой. Удаление селезёнки (спленэктомия) при наследственном сфероцитозе улучшает состояние больных, так как предотвращает разрушение сфероцитов в селезёнке.
4. Мегалобластная (макроцитарная) анемия развивается при дефиците фолиевой
кислоты или витамина В12.
Фолиевая кислота в виде кофермента (Н4-фолата) участвует в синтезе нуклеотидов.
Недостаток фолиевой кислоты приводит к снижению скорости синтеза ДНК в быстроделящихся клетках (в т.ч. в предшественниках эритроцитов). Клетки дольше пребывают в интерфазе, синтезируя гемоглобин, и становятся крупнее. Кроме того, из-за недостатка нуклеотидов они реже делятся, и количество эритроцитов снижается, а крупные мегалобласты быстрее разрушаются. Всё это в конечном итоге приводит к развитию анемии.
Аналогичная симптоматика развивается при недостатке в организме витамина В12.
Этот витамин участвует в переносе метальной группы с N5-метил-Н4-фолата на гомоцистеин с образованием метионина и Н4-фолата. Недостаточность витамина В12 приводит к накоплению N5-метил-Н4-фолата в клетках. Дефицит Н4-фолата приводит к нарушению деления клеток и развитию анемии
Вопрос 104
Биосинтез гема. Схема процесса, химизм первых двух реакций, место протекания. Регуляция активности ферментов АЛК-синтазы и АЛК-дегидратазы. Источники железа для синтеза гема, всасывание железа, транспорт в крови, депонирование
Гем синтезируется во всех тканях, но с наибольшей скоростью в костном мозге (для образования гемоглобина в ретикулоцитах) и печени (для образования Цитохрома Р450)
Первая реакция синтеза гема – образование 5-аминолевулиновой к-ты
Катализирует: 5-аминолевулинатсинтаза ПФ-зависимая (АЛК-синтаза)
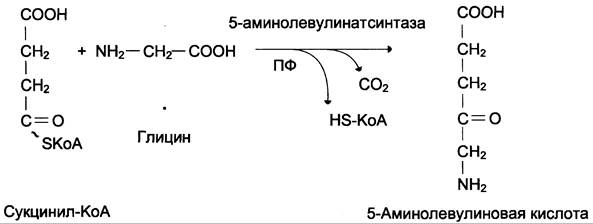
Из митохондрий 5-аминолевулиновая к-та поступает в цитоплазму, где
происходит 2 реакция - соединение 2 молекул 5-аминолевулиновой к-ты с образованием
порфобилиногена под действием аминолевулинатдегидрогеназы (АЛК-дегидрогеназа)
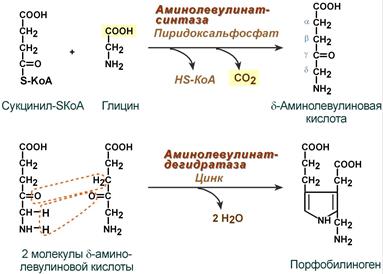
Далее в ходе последовательных реакций обр-ся гем (их химизм не нудно знать)

Регуляция активности АЛК-синтазы и АЛК-дегидрогеназы: