

Вопрос 85.
Обмен фенилаланина и тирозина. Особенности обмена тирозина в разных тканях. Синтез катехоламинов, меланинов, йодтиронинов. Наследственные биохимические блоки в распаде фенилаланина и тирозина: паркенсонсизм, фенилкетонурия, алкаптонурия, альбинизм, диагностика и лечение.
Фенилаланин – незаменимая аминокислота, в клетках животных
не синтезируется её бензольное кольцо.
Тирозин – условно заменимая аминокислота, образуется из фенилаланина.
Основной путь метаболизма начинается с гидроксилирования фенилаланина, в результате образуется тирозин. Обмен фенилаланина и тирозина катализируют оксигеназы, которые используют О2 и кофермент-донор водорода тетрагидробиоптерин (Н4БП).
Для катализа оксигеназам необходимы кофакторы – Fe2+ или гем.
Монооксигеназы – один атом О2, присоединяют к продукту реакции, другой используют для образования Н2О.
Диоксигеназы – оба атома О2 используют для образования р-ии.
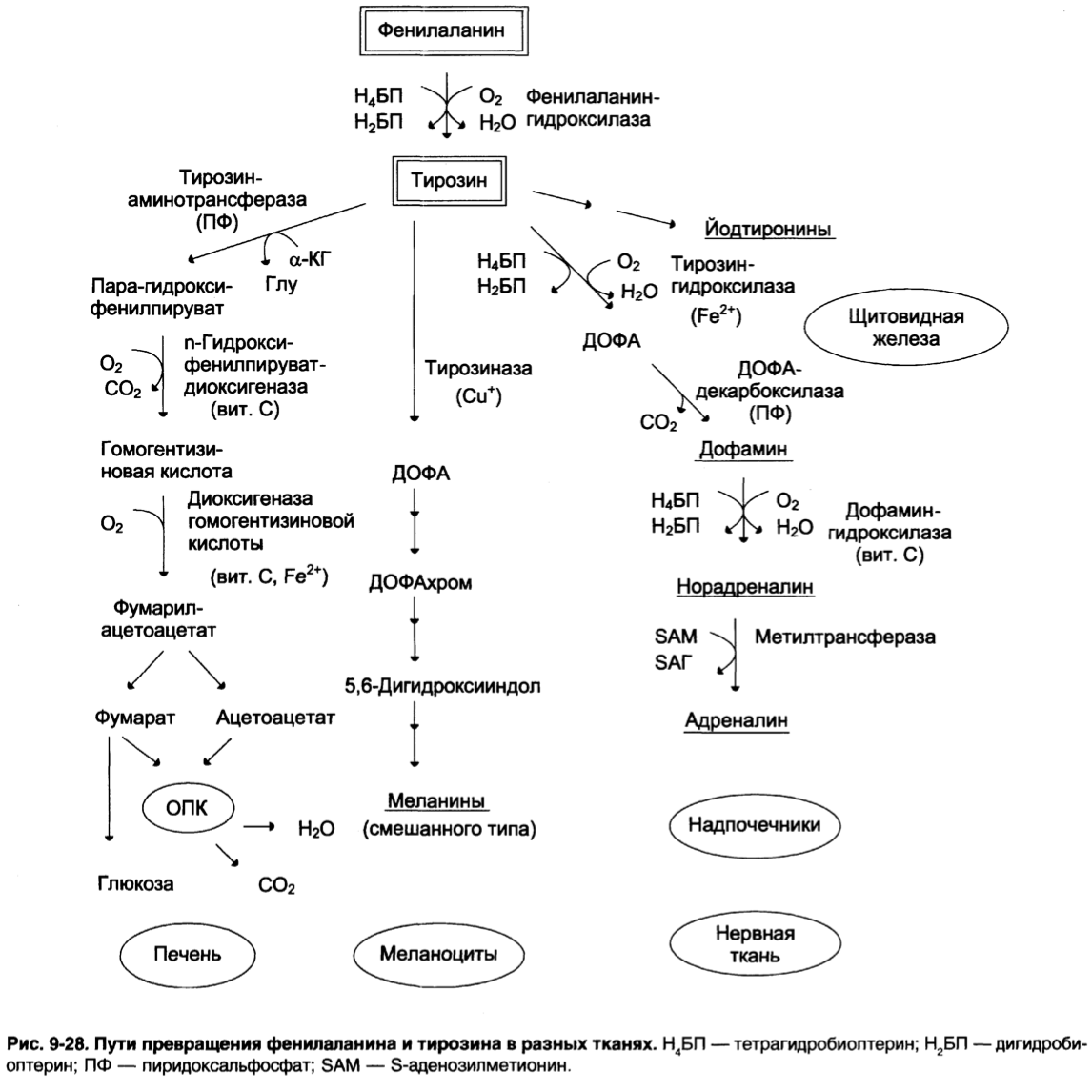
Особенности обмена тирозина в разных тканях:
Кроме использования в синтезе белков, тирозин в разных тканях явл.
предшественником катехоламинов, тироксина, меланинов и распадается до СО2 и Н2О
1. Катаболизм тирозина в печени: происходит до конечных продуктов –
фумарата и ацетоацетата
1 – Тирозинаминотрансфераза (ПФ-зависимая)
2 – П-гидроксифенилпируватдиоксигеназа (кофакторы – вит. С и Fe2+)
Происходит декарбоксиллирование, гидроксилирвоание аромат. кольца
и миграция боковой цепи
3 – Диоксигеназа гомогентизиновой к-ты (кофермент Fe2+)
4 – Фумарилоацетоацетатгидролаза

2. В меланоцитах (пигментные клетки): они находятся в составе волос,
в сетчатке глаза, определяют цвет кожи
Тирозиназа – ф., превращающий тирозин в ДОФА в клетках меланоцитов
(кофактор – Cu).
Из ДОФА затем будут многоступенчато синтезироваться мелнины.
Эумеланины (черного или коричневого цвета)
Феомеланины (желтого или красновато-коричневые)
3. В щитовидной железе: синтезируются йодтиронины (тироксин и трийодтрионин) –
это йодированные остатки тирозина, которые попадают в клетки щитовидной железы через базальную мембрану
4. В надпочечниках и нервной ткани (синтез катехоламинов):
В мозговом веществе надпочечников и нервной ткани - предшественник катехоламинов.
Катализируются ферментами:
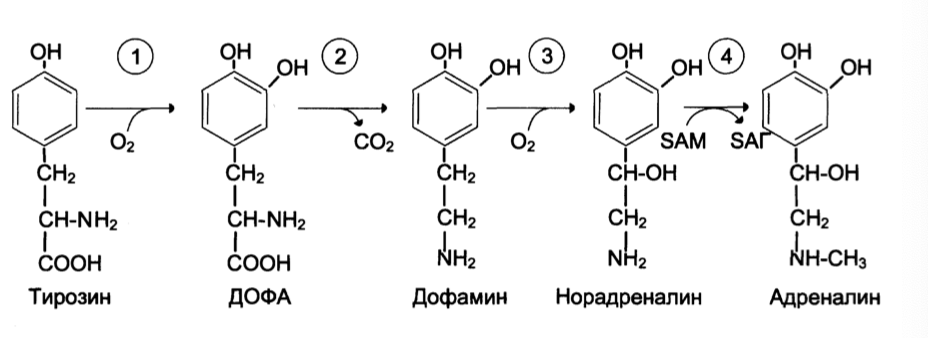
Тирозингидроксилаза (1) – Fe зависимый фермент, является регулятором
и определяет скорость синтеза.
Активность его изменяется в результате: аллостерической регуляции
(ингибитор – норадреналин).
Фософорилирования/дефосфорилирования
ДОФА-декарбоксилаза (2) (кофермент ПФ) катализирует образование дофамина, который при участии дофамингидроксилазы (3) превращается в норадреналин
Фенилэтаноламин-N-метилтрансфераза (4) катализирует метилирование норадреналина в адреналин
Фенилкетонурия (ФКУ) – наследственное заболевание, связанное с нарушением
катаболизма фенилаланина. Сопровождается гиперфенилаланинемией и повышением в крови и моче содержания фенилпирувата, фенилацетата и тд
Дефект фенилаланингидроксилазы приводит к заболеванию ФКУ:
Классическая ФКУ - следствие мутаций в гене фенилаланингидроксилазы, которые приводят к снижению активности фермента или полной его инактивации.
Концентрация фенилаланина повышается в крови в 20-30 раз
В моче – в 100-300 раз по сравнению с нормой
Концентрация фенилпирувата и фениллактата в моче достигает 300-600 мг/дл
при полном отсутствии в норме.
Проявления: нарушение умственного и физического развития, судорожный синдром, нарушение пигментации вследствие токсического действия на мозг высоких конц. фенилаланина, фенилпирувата, фениллактата. Также они тормозят транспорт тирозина и триптофана через ГЭБ и тормозят синтез нейромелдиаторов.
При отсутствии лечения больные не доживают до 30 лет.
Вариантная ФКУ – следствие мутаций в генах, контролирующих метаболизм Н4БП.
Клинические проявления – близкие с проявлениями классической ФКУ.
Частота заболевания – 1-2 случая на 1 млн новорождённых.
Нарушение умственного и физического развития у детей, больных ФКУ, можно предотвратить диетой с очень низким содержанием или полным исключением фенилаланина.
Тирозинемии – нарушения катаболизма тирозина в печени.
Тирозинемия типа 1 (тирозиноз): причиной явл. дефект фермента
фумарилацетоацетатгидролазы, катализирующего расщепление фумарилацетоацетата на фумарат и ацетоацетат
Острая форма – характерна для новорождённых.
Клинические проявления: диарея, рвота, задержка в развитии. Без лечения дети погибают в возрасте 6-8 мес из-за недостаточности печени.
Хроническая форма – сходные, но менее выраженные симптомы. Гибель в
возрасте 10 лет. Лечение: диета с пониженным содержанием тирозина и фенилаланина.
Тирозинемия типа II – причиной явл. дефект фермента тирозинаминотрансферазы.
Концентрация тирозина в крови больных повышена. Проявляется поражением глаз и кожи, умеренная умственная отсталость, нарушение координации движений.
Тирозинемия новорождённых (кратковременная) – повышенная концентрация п-
гидроксифенилацетата, тирозина и фенилаланина. Лечение: бедная белком диета и витамин С.
Алкаптонурия ("чёрная моча") – причиной явл. дефект диоксигеназы
гомогентизиновой кислоты
Проявления: потемнения мочи на воздухе, пигментация соединительной ткани, артрит.
Альбинизм – причиной явл. врождённый дефект тирозиназы (она катализирует
превращение тирозина в ДОФА в меланоцитах) = нарушается синтез пигментов меланинов. Проявления: отсутствие пигментации кожи и волос, часто снижена острота зрения, светобоязнь.