

Ацилирование и алкилирование аминов
.pdfСПБГУАП группа 4736 https://new.guap.ru/i03/contacts
Обычно, для получения соли к амину прибавляют небольшой избыток кислоты, например разбавленной серной, концентрированной или разбавленной соляной или бромистоводородной кислоты. Если при этом соль не выпадает, раствор упаривают на водяной бане или в вакуум-эксикаторе до начала кристаллизации. Если галоидоводородные соли плохо кристаллизуются из водного раствора, их получают другим путем, а именно пропусканием сухого галоидоводорода в раствор амина в бензоле, хлороформе или эфире. Этот метод особенно удобен для получения галоидоводородных солей алкиланилинов и диалкиланилинов, а также аминов, соли которых легко гидролизуются водой. При пропускании сухого хлористого водорода в раствор диалкиланилина в сухом эфире до насыщения соответствующие солянокислые соли легко выделяются в кристаллическом состоянии. При этом следует тщательно предохранять реакционную смесь от доступа влаги. Солянокислые соли низших алкиланилинов лучше всего получать в бензольном растворе. Впрочем, в случае алкиланилинов, содержащих сравнительно большие алкильные радикалы, этот способ не дает таких удовлетворительных результатов.
Большинство аминов образует хорошо кристаллизующиеся пикраты, которые могут служить для идентификации аминов или для выделения их из смесей. Обычно, пикраты получаются смешением обоих компонентов в подходящем растворителе, выбор которого определяется сравнительной растворимостью в нем пикриновой кислоты, пикрата и амина. Менее удобно пользоваться для этой цели реакцией обмена. Пикролоновая кислота также применяется для идентификации аминов, особенно в тех случаях, когда пикриновая кислота не дает удовлетворительных результатов. Соли пикролоновой кислоты обычно труднее растворимы, чем пикраты, и обладают более высокой температурой плавления. Этот способ применяется главньм образом для идентификации простейших алифатических производных гидроксиламина, производных морфолина и некоторых алкалоидов. Кроме того, для идентификации аминов также применяется имидазолдикарбоновая кислота.
Ароматические амины образуют продукты присоединения с ди- и тринитросоединениями. Эти продукты также иногда служат для идентификации аминов. Некоторые амины при действии 70% водной хлорной кислоты дают хорошо кристаллизующиеся соли, которые могут служить для их выделения и идентификации [3,4].
Реакции ацилирования
Ацилирование - введение ацильной группы (ацила) RC в молекулу органического соединения путем замещения атома водорода. В широком смысле ацилирование это замещение любого атома или группы атомов на ацила. В зависимости от атома к которому присоединяют ацил различают C-, N-, O-, S - ацилирование.
Реакции ацилирования обладают очень многими полезными свойствами. Они позволяют вести в молекулу функциональную группу C=O
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
путем реакций присоединения либо замещения, не подвергая исходную молекулу окислению (восстановлению). Таким образом, можно получать соединения различных классов: а) амиды; б) сложные эфиры; в) ангидриды карбоновых кислот; г) кетоны и другие полезные соединения. Неудивительно, что реакции ацилирования находят широкое применение в промышленности и в химических исследованиях. В своей курсовой работе я рассмотрю три наиболее важных типа реакций ацилирования C- ацилирование, O-ацилирование и N-ацилирование.
Ацилирование аминов ацилгалогенидами Реакции аминодегалогенирования наиболее часто используются для
синтеза амидов. Действие аммиака или аминов на ацилгалогениды представляет собой общий метод синтеза амидов (Рис. 13):
Рис. 13. Общий метод синтеза амидов
Реакция сильно экзотермична и требует тщательного контроля, обычно охлаждением или разбавлением. При использовании аммиака получают незамещенные амиды, из первичных аминов получают N-замещенные амиды, а из вторичных аминов - N, N-дизамещенные амиды. Аналогично можно ацилировать ариламины. В некоторых случаях для связывания выделяющейся галогеноводородной кислоты добавляют водный раствор щелочи. Такая реакция носит название метода Шоттена-Баумана.
Гидразин и гидроксиламин также реагируют с ацилгалогенидами, давая соответственно гидразиды RCONHNH2 и гидроксамовые кислоты RCONHOH; эта реакция часто используется для синтеза данных соединений. Если вместо ацилгалогенида взять фосген, то как ароматические, так и алифатические первичные амины дают хлороформамиды ClCONHR, которые теряя HCl, превращаются в изоцианаты RNCO. Это один из наиболее распространенных методов синтеза изоцианатов.
Тиофосген при аналогичной обработке дает изотиоцианаты. Фосген в этой реакции можно заменить более безопасным трихлорометилхлороформиатом. При действии первичных аминов на хлороформиаты ROCOCl получаются карбаматы ROCONHR’. Примером этой реакции служит защита аминогруппы в аминокислотах и пептидах действием карбобензоксихлорида (Рис. 14):
Рис. 14. Защита аминогруппы в аминокислотах и пептидах действием карбобензоксихлорида
Аминогруппы вообще часто защищают превращением в более устойчивые - амидные. Взаимодействие ацилгалогенидов с нитридом лития дает N, N-диациламиды (триациламины).
Ацилирование аминов ангидридами По механизму и диапазону применимости реакция амино-
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
деацилоксизамещения может быть проведена с участием аммиака, первичных или вторичных аминов (Рис. 15)
Рис. 15. Реакция аминодеацилоксизамещения Однако при использовании аммиака и первичных аминов получаются
также и имиды, в которых с атомом азота связаны две ацильные группы. Это происходит особенно легко в случае циклических ангидридов, из которых образуются циклические амиды (Рис. 16):
Рис. 16. Реакция получения имидов
Второй стадией этой реакции, которая намного медленнее первой, является атака атома азота амидной группы на карбоновую кислоту.
Ацилирование аминов карбоновыми кислотами При обработке карбоновых кислот аммиаком или аминами получаются
соли. Соли, полученные из аммиака, а также первичных и вторичных аминов в результате пиролиза дают амиды, но этот метод менее удобен, чем реакции аминов с ангидридами, ацилгалогенидами и сложными эфирами, и редко используется в препаративных целях [5].
Хотя и взаимодействие кислот с аминами не приводит непосредственно к амидам, можно добиться чтобы эта реакция шла с хорошим выходом при комнатной или немного повышенной температуре (Рис. 17):
Рис. 17. Реакция получения амидов
Кислоты можно превратить в амиды также нагреванием с амидами других карбоновых кислот (обмен), сульфоновых или фосфиновых кислот или действием трис(алкиламино) боранов [B(NHR’) 3] или трис(диалкиламино) боранов [B(NR’2) 3] (Рис. 18):
Рис. 18. Превращение кислот в амиды
Ацилирование аминов сложными эфирами Превращение сложных эфиров в амиды - полезный метод синтеза
незамещенных, N-замещенных и N, N-дизамещенных амидов из соответствующих аминов (Рис. 19):
Рис. 19. Превращение сложных эфиров в амиды
Реакцию можно проводить с алкильными или ароматическими группами R и R’. Особенно хорошей уходящей группой является n- нитрофенильная. Эта реакция весьма ценна, так как многие сложные эфиры легкодоступны или сравнительно легко получаются даже в тех случаях, когда этого нельзя сказать о соответствующем ангидриде кислоты или ацилгалогениде. Как и по реакции с ацилгалогенидами, этим методом из сложных эфиров можно синтезировать гидразиды и гидроксамовые кислоты
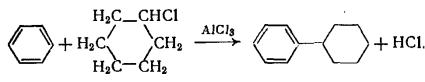
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
действием гидразина и гидроксиламина соответственно. И гидразин, и гидроксиламин взаимодействуют быстрее, чем аммиак или первичные амины. Вместо сложных эфиров часто используют фенилгидразиды, получаемые из фенилгидразина.
Остаётся добавить, что реакция образования гидроксамовых кислот. которые в присутствии трёхвалентного железа дают окрашенные комплексы, часто используется как тест на сложные эфиры.
Ацилирование аминов амидами Это реакция обмена, и ее обычно проводят с солью амина. Уходящей
группой служит, как правило, NH2, а не NHR или NR2; в качестве реагентов наиболее широко применяются первичные амины (в виде солей).
Для образования комплекса с уходящим аммиаком можно добавлять BF3. Эту реакцию часто применяют для получения замещенных производных мочевины из самой мочевины (Рис. 20):
Рис. 20. Ацилирование аминов амидами
Диметилформамид можно превратить в другие формамиды продолжительным нагреванием с первичным или вторичным амином (Рис.
21) [10]:
Рис. 21. Превращение диметилформамида в другие формамиды
Реакции алкилированияалкилирование нередко классифицируют как аммонолиз (или аминолиз) органических соединений).
Алкилирование по атомам других элементов (Si-, Pb-, AIалкилирование) представляет собой важнейший путь получения элемент- и металлорганических соединений, когда алкильная группа непосредственно связывается с гетероатомом (Рис. 22):
|
Cu |
|
RCI + Si |
|
R2SiCI2 |
|
C2H5CI + 4PbNa → Pb(C2H5)4 + 4NaCI + 3Pb C3H6 + AI + 1,5H2 → Al(C3H7)3
Рис. 22. Реакции алкилирования Другая классификация реакций алкилирования основана на различиях в
строении алкильной группы, вводимой в органическое или неорганическое соединение. Она может быть насыщенной алифатической (этильной и изопропильной) или циклической. В последнем случае реакцию иногда называют циклоалкилированием (Рис. 23):
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
Рис. 23. Реакция циклоалкилированием
При введении фенильной или вообще арильной группы образуется непосредственная связь с углеродным атомом ароматического ядра (арилирование) (Рис. 24):
H5CI + NH3 → C6H5NH2 + HCI
Рис. 24. Арилирование
В алкильную группу может входить ароматическое ядро или двойная связь, и, если последняя достаточно удалена от реакционного центра, реакция мало отличается от обычных процессов алкилирования (Рис. 25):
=CH-CH2CI + RNH2 → RNHCH2-CH=CH2 + HCI
Рис. 25. Реакция алкилирования
Однако введение винильной группы (винилирование) занимает особое место и осуществляется главным образом при помощи ацетилена (Рис. 26):
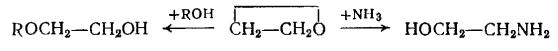
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
ROH + CH≡CH
HO
ROCH=CH2-COOH + CH≡CH
2
Zn CH3-COO-CH=CH2
Рис. 26. Реакция винилирования ацилирование алкилирование амин
Наконец, алкильные группы могут содержать различные заместители, например атомы хлора, гидрокси-, карбокси-, сульфокислотные группы (Рис.
27):
H5ONa + CICH2-COONa → C6H5O-CH2-COONa + NaCI+ HOCH2-
CH2SO2ONa → ROCH2-CH2SO2ONa + H2O
Рис. 27. Структура алкильных групп
Важнейшей из реакций введения замещенных алкильных групп является процесс β-оксиалкилирования (в частном случае оксиэтилирование), охватывающий широкий круг реакций оксидов олефинов (Рис. 28):
Рис. 28. Оксиэтилирование
Алкилирующие агенты и катализаторы Все алкилирующие агенты по типу связи, разрывающейся в них при
алкилировании, целесообразно разделить на следующие группы:
. Ненасыщенные соединения (олефин и ацетилен), у которых
происходит разрыв |
|
-электронной связи между атомами углерода; |
|
. Хлорпроизводные с достаточно подвижным атомом хлора, способным замещаться под влиянием различных агентов;
. Спирты, простые и сложные эфиры, в частности оксиды олефинов, у которых при алкилировании разрывается углерод-кислородная связь.
Олефины (этилен, пропилен, бутены и высшие) имеют первостепенное значение в качестве алкилирующих агентов. Ввиду дешевизны ими стараются пользоваться во всех случаях, где это возможно. Главное применение они нашли для С-алкилирования парафинов и ароматических соединений. Они неприменимы для N-алкилирования и не всегда эффективны при S- и O-алкилировании и синтезе металлорганических соединений [7].
Алкилирование олефинами в большинстве случаев протекает по ионному механизму через промежуточное образование карбокатионов и катализируется протонными и апротонными кислотами. Реакционная способность олефинов при реакциях такого типа определяется их склонностью к образованию карбокатионов:
RCH CH 2 H RCH CH3 (4)
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
Это означает, что удлинение и разветвление цепи углеродных атомов в олефине значительно повышает его способность к алкилированию:
=CH2 < CH3-CH=CH2 < CH3-CH2-CH=CH2 < (CH3)2C=CH2 (5)
В ряде случаев алкилирование олефинами протекает под влиянием инициаторов радикально-цепных реакций, освещения или высокой температуры. Здесь промежуточными активными частицами являются свободные радикалы. Реакционная способность разных олефинов при таких реакциях значительно сближается.
Хлорпроизводные являются алкилирующими агентами наиболее широкого диапазона действия. Они пригодны для С-, О-, S- и N- алкилирования и для синтеза большинства элементо- и металлорганических соединений. Применение хлорпроизводных рационально для тех процессов, в которых их невозможно заменить олефинами или когда хлорпроизводные дешевле и доступнее олефинов.
Алкилирующее действие хлорпроизводных проявляется в трех различных типах взаимодействий: в электрофильных реакциях, при нуклеофильном замещении и в свободно-радикальных процессах. Механизм электрофильного замещения характерен для алкилирования по атому углерода, но, в отличие от олефинов, реакции катализируются только апротонными кислотами (хлориды алюминия, железа). В предельном случае процесс идет с промежуточным образованием карбокатиона:
|
|
|
R. AICI |
|
RCI AICI |
R |
CI AI CI |
(6) |
|
3 |
|
3 |
4 |
в связи, с чем реакционная способность алкилхлоридов зависит от поляризации связи C-CI или от стабильности карбокатионов и повышается при удлинении и разветвлении алкильной группы:
CH |
3 |
CH |
CI (CH |
) |
CHCI (CH |
) |
CCI |
|
2 |
3 |
2 |
3 |
3 |
|
(7)
При другом типе реакций, характерном для алкилирования по атомам кислорода, серы и азота, процесс состоит в нуклеофильном замещении атома хлора. Механизм аналогичен гидролизу хлорпроизводных, причем реакция протекает в отсутствие катализаторов:
|
|
|
|
|
|
|
|
RCI : NH |
|
R NH |
CI |
RNH |
|
HCI |
|
3 |
|
3 |
|||||
|
3 |
|
|
|
|
(8)
Реакционная способность хлорпроизводных изменяется в данных процессах таким же образом, как при гидролизе, а именно:
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
ArCH2CI > CH2=CH-CH2CI > AIkCI > ArCI (9) перв-AIkCI > втор-AIkCI > трет-AIkCI (10)
Целый ряд процессов алкилирования хлорпроизводными протекает по свободно-радикальному механизму. Это особенно характерно для синтезов элементо- и металлорганических соединений, когда свободные радикалы образуются за счет взаимодействия с металлами [6]:
PbNa + 4C2H5CI → 4Pb + 4NaCI + 4C2H |
. |
|
5 |
||
|
(11)
→ 4NaCI + Pb(C2H5)4 + 3Pb
Ацилирование и алкилирование по Фриделю-Крафтсу Ацилирование по Фриделю-Крафтсу Введение ацильной группы в ароматическое кольцо с помощью
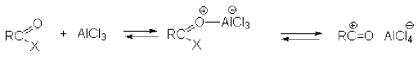
ацилирующего агента и кислоты Льюиса называют ацилированием по Фриделю-Крафтсу. Ацилирующими агентами обычно являются галогенангидриды и ангидриды кислот в присутствии галогенидов алюминия, трифторида бора или пентафторида сурьмы в качестве кислот Льюиса. Ацилгалогениды и ангидриды кислот образуют с кислотой Льюиса донорно-акцепторные комплексы состава 1:1 и 1:2. Спектральными методами было установлено, что хлорид алюминия, трифторид бора и пентафторид сурьмы координируются по карбонильному атому кислорода, так как он более основен чем соседний атом хлора. Электрофильным агентом в реакции ацилирования ароматических соединений является либо этот донорноакцепторный комплекс, либо катион ацилия, образующийся при его диссоциации (Рис. 29).
Рис. 29. Реакция ацилирования по Фриделю-Крафтсу Можно полагать, что медленной стадией реакции является атака одного
из трех электрофилов на арен, приводящая к σ-комплексу. Эффективность этих ацилирующих частиц зависит от природы субстрата, ацилгалогенида и растворителя, а также от количества взятого катализатора.
При ацилировании аренов ацилгалогенидами, катализируемом хлоридом или бромидом алюминия в полярных апротонных растворителях (нитробензоле, нитрометане и др.), ацилирующим агентом является катион ацилия, тогда как в малополярной среде (хлористом метилене, дихлорэтане или тетрахлорэтане) в реакции принимает участие донорно-акцепторный комплекс. Природа ацилгалогенида также оказывает влияние на образование и стабильность солей ацилия. Механизм реакции ацилирования аренов по Фриделю-Крафтсу под действием донорно-акцепторного комплекса (Рис.
30):

СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
Рис. 30. Механизм реакции ацилирования аренов по Фриделю-Крафтсу
Ароматический кетон представляет собой более сильное основание Льюиса, чем ацилгалогенид и образует стабильный комплекс с AlCl3 или другой кислотой Льюиса. Поэтому для ацилирования ароматических соединений ацилгалогенидами требуется несколько больше эквимолярного количества катализатора, а при ацилировании ангидридами кислот два моля катализатора (т.к. они содержат два карбонильных атома кислорода). Кетон выделяют, разлагая его комплекс с AlCl3 водой или соляной кислотой.
Ацилирование по Фриделю-Крафтсу полностью лишено тех недостатков, которые присущи реакции алкилирования. При ацилировании вводится только одна ацильная группа, поскольку ароматические кетоны не вступают в дальнейшую реакцию (так же, как и другие арены, содержащие сильные электроноакцепторные группы: NO2, CN, COOR). Еще одним преимуществом этой реакции по сравнению с алкилированием является отсутствие перегруппировок в ацилирующем агенте. Кроме того, для ацилирования не характерны реакции диспропорционирования продуктов реакции [4,7].
Алкилирование по Фриделю-Крафтсу Реакция Ш.Фриделя-Дж.Крафтса (1877 г.) представляет собой удобный
метод прямого введения алкильной группы в ароматическое кольцо. Алкилирование ароматических соединений осуществляется под действием алкилгалогенидов, только в присутствии в качестве катализатора подходящей кислоты Льюиса: AlBr3, AlCl3, GaBr3, GaCl3, BF3, SbF5, SbCl5, FeCl3, SnCl4, ZnCl2 и др. (Рис. 31):
Рис. 31. Алкилирование ароматических соединений
Наиболее активными катализаторами являются безводные сублимированные бромиды алюминия и галлия, пятифтористая сурьма, хлориды алюминия и галлия, менее активны галогениды железа (III), SbCl5, к малоактивным катализаторам относятся SnCl4 и ZnCl2. В целом активность кислот Льюиса, как катализаторов алкилирования бензола, уменьшается в ряду
СПБГУАП группа 4736 https://new.guap.ru/i03/contacts
>GaBr3>AlCl3>GaCl3>FeCl3>SbCl5>TiCl4>BF3>BCl3>SnCl4>SbCl3.