

Биология Ярыгин 2003
.pdfЯвлением ГИ объясняется, например, избирательная инактивация у млекопитающих отцовской Х-хромосомы в клетках провизорных органов (см. гл. 7.5.4.). В клетках самого зародыша имеет место равновероятная инактивация отцовской и материнской Х-хромосом (см. рис. 3.78).
Таким образом, следствием ГИ (дифференциальной маркировки в гаметогенезе родителей и последующей избирательной инактивации у потомков участков хромосом) является функциональная неравноценность в генотипе потомка аллелей разного родительского происхождения.
Связь этиологии ряда наследственных заболеваний с феноменом ГИ может быть прослежена на разных уровнях организации генетического материала.
На геномном уровне организации наследственного материала доказательством роли ГИ в патологии служит различное фенотипическое проявление триплоидных состояний при разном соотношений гаплоидных наборов отцовского и материнского происхождения.
У диандрических триплоидов (соотношение числа гаплоидных наборов отца и матери 2:1) и у дигенических триплоидов (соотношение 1:2) патологические отклонения в развитии плаценты и собственно зародышевых тканей проявляются по-разному. Это свидетельствует о неравноценности функционирования гаплоидных наборов отца и матери в тканях зародыша и плаценты (см, разд. 7.5.4 и 7.6.1).
Связь феномена ГИ с патологией на уровне отдельных хромосом можно проследить в случае однородительской дисомии (ОРД), при которой происходит удвоение хромосомы одного из родителей при утрате гомологичной хромосомы другого родителя.
Воснове возникновения ОРД лежит нарушение процессов гаметогенеза. При нерасхождении сестринских хроматид в анафазе II мейоза появляются гаметы, в галлоидном наборе которых присутствуют две генетически идентичные хромосомы (изодисомия).
Вслучае нерасхождения гомологичных хромосом в анафазе I мейоза образуются гаметы, в гаплоидном наборе которых имеется пара гомологичных, генетически неидентичных хромосом (гетеродисомия). В обоих случаях гаметы данного индивида дисомны по одной из хромосом.
При оплодотворении дисомных гамет нулисомными по той же хромосоме подовыми клетками происходит комплемеитация гамет, приводящая к
возникновению нормального диплоидного кариотипа зиготы. Однако в генотипе такой зиготы присутствует двойной набор генов данной хромосомы, происходящих от одного, а не от обоих родителей.
Иногда оплодотворение дисомных гамет нормальными половыми клетками сопровождается «коррекцией трисомии» в результате потери сверхчисленной хромосомы. Если при этом сохраняются две хромосомы, пришедшие от одного родителя, то наблюдается явление ОРД.
Наконец, состояние ОРД по отдельным локусам хромосом может возникать в
271
medwedi.ru
результате соматической рекомбинации — кроссинговера между хроматидами гомологичных хромосом, происходящего в соматических клетках (см. рис 3.73).
Когда хромосома не содержит импринтированных участков, при ОРД по данной хромосоме может не наблюдаться аномалий фенотипа. Исключением может быть проявление аутосомно-рецессивного заболевания как результат гомозиготизации по рецессивному аллелю при изодисомии.
Если хромосома содержит импринтированные участки, то при возникновении однородительской дисомии локализованные в них аллели могут быть либо экспрессированы, либо инактивированы в зависимости от родительского происхождения ОРД. Это может стать причиной возникновения патологических отклонений в развитии организма. Фенотипическое проявление при ОРДмат и ОРДотц может быть сходным или прямо противоположным. Возможен летальный эффект уже на ранних сроках развития.
В настоящее время эффект импринтинга установлен достаточно определенно для четырех хромосом человека 15, 11, 7, 14. Так в проксимальном отделе длинного плеча 15-й хромосомы имеется район, подверженный импринтингу. Мутации, связанные с микроделециями в этом районе, приводят к развитию у человека синдрома Прадера — Вилли, при котором у пациентов наблюдается умственная отсталость, мышечная гипотония, сильное ожирение, гипогонадизм, низкий рост, акромикрия (непропорционально малые размеры дистальных отделов конечностей). В настоящее время описано более 30 случаев синдрома Прадера —Вилли, когда у пациентов определяется ОРДмат 15. Считается, что ОРДмат 15 является причиной 20—25% всех случаев этого синдрома. Большая же часть остальных случаев заболевания связана с делецией сегмента 15qll — ql3 отцовской хромосомы. Указанный пример свидетельствует об активной экспрессии соответствующего участка 15-й хромосомы исключительно отцовского происхождения. В материнской же хромосоме он метилирован и репрессирован.
Делеция другого участка, также расположенного в сегменте 15qll — ql3, но в 15-й хромосоме материнского происхождения, в 70% случаев приводит к развитию синдрома Энгельмана (синдрома «счастливой куклы»), характеризующегося глубокой умственной отсталостью с резкими судорожными движениями и неадекватной счастливой улыбкой. В 2% случаев этот синдром обусловлен ОРДотц15.
Из сказанного выше следует, что в проксимальном районе длинного плеча 15- й хромосомы имеются близкорасположенные и противоположно импринтированные локусы, отвечающие за возникновение фенотипически различных синдромов Прадера — Вилли и Энгельмана.
Таким образом импринтироваться могут участки хромосом разного родительского происхождения, что и определяет нетрадиционное наследование многих патологических состояний, обусловленных мутациями локусов, подверженных импринтингу.
Митохондриальные болезни. Начиная с конца 80-х годов XX века получены
272
убедительные доказательства связи некоторых видов наследственной патологии у человека с мутациями митохондриальной ДНК (см. гл. 4.1) В зависимости от типа мутаций митохондриальные болезни разделяют на 4 группы:
а) болезни, вызванные точковыми мутациями, приводящими к замене консервативных аминокислот в собственных белках митохондрий. К ним относятся пигментный ретинит и нейроофтальмопатия Лебера, при которой наступает двусторонняя потеря зрения. Выраженность клинических признаков у больных этими заболеваниями коррелирует с количеством мутантной мтДНК, которое у разных больных может варьировать от 5 до 100% всей мтДНК;
б) болезни, вызванные мутациями в генах т-РНК, приводящими к многочисленным дегенеративным заболеваниям с различной степенью тяжести клинических проявлений, коррелирующей с количеством мутантной мтДНК;
в) болезни вызванные делениями и дупликациями участков митохондриалъных генов. У человека описано тяжелое заболевание молодого и среднего возраста — отсроченная кардиопатия, при которой обнаружены делеции мтДНК кардиоцитов. Заболевание носит семейный характер. В ряде случаев предполагается Х- сцепленное наследование, что позволяет думать о существовании ядерного гена, мутация которого вызывает делению до 50% мтДНК кардиоцитов;
г) болезни, вызванные снижением числа копий мтДНК, что является следствием определенных мутаций. К данной группе относятся летальная инфантильная дыхательная недостаточность и синдром молочнокислого ацидоза, при которых число копий мтДНК снижается до 1—2% от нормы. Снижение содержания мтДНК в клетках различных органов приводит к развитию миопатий, нефропатий, печеночной недостаточности и т.д. вследствие ослабления синтеза белков, кодируемых мтДНК.
Изменения в ДНК митохондрий сопровождаются нарушением их функций, связанных с клеточным дыханием. Это определяет характер и степень тяжести клинических проявлений митохондриалъных болезней.
Выдвинута также гипотеза о том, что накопление спонтанно возникающих мутаций мтДНК является звеном механизмов старения и развития дегенеративных процессов у человека.
Болезни экспансии тринуклеотвдных повторов с явлением антиципации.
Под генетической антиципацией (или упреждением) понимается более раннее проявление и возрастание тяжести симптомов наследственного заболевания в последующих поколениях родословной. Антиципация реально проявляется при определенных видах моногенной неврологической патологии, а также при некоторых мультифакториальных заболеваниях.
В начале 90-х годов XX века при исследовании ряда тяжелых неврологических заболеваний были обнаружены «динамические» мутации с экспансией (резким увеличением числа копий) тринуклеотидных повторов у индивидов в последующих поколениях родословной. Развивающиеся в результате таких мутаций наследственные заболевания характеризуются четко выраженным проявлением антиципации.
273
medwedi.ru
Феномен экспансии числа тринуклеотидных повторов был впервые обнаружен при исследовании синдрома Мартина—Белла или синдрома фрагильной (ломкой) Х- хромосомы, основным фенотипическим проявлением которого является умственная отсталость. Синдром ломкой Х-хромосомы характеризуется довольно широкой распространенностью в популяции (1:1000) и необычным характером наследования. Лишь у 80% мужчин-носителей мутантного локуса имеются клинические и цитогенетические признаки заболевания. 20% носителей как клинически, так и цитогенетически нормальны, но после передачи мутации всем своим дочерям они могут иметь пораженных внуков. Неэкспрессируемый мутантный ген в таком случае становится экс-прессируемым в последующих поколениях.
Таким образом мутантный ген при синдроме ломкой Х-хромосомы может существовать в двух формах, отличающихся по своей пенетрантности. Одна — фенотипически не проявляющаяся — премутация, которая при прохождении через женский мейоз превращается в другую форму — полную мутацию. При таком необычном способе наследования и фенотипического проявления мутантного гена, отличном от классического Х-сцепленного наследования, обнаруживается феномен антиципации — более тяжелое проявление заболевания в последующих поколениях.
В основе клинических проявлений и цитологической нестабильности в локусе, ответственном за синдром ломкой Х-хромосомы, лежит многократное увеличение повторов тринуклеотида ЦГГ. В норме число повторов колеблется от 5 до 50. Премутация — неэкспрессируемая форма — характеризуется увеличением числа повторов до 50—200. Возрастание числа повторов тринуклеотида ЦГГ свыше 200 приводит к клинической манифестации заболевания и цитогенетическому проявлению ломкой Х-хромосомы. Как правило, у пораженных лиц наблюдается также аномальное метилирование ДНК, приводящее к репрессированию гена.
Интересно, что переход от состояния премутации к полной мутации возникает при передаче от матери, причем экспансия ЦГГ-повторов значительно выше при передаче от матери к сыну, чем от матери к дочери.
Антиципация, характерная для синдрома ломкой Х-хромосомы, объясняется четкой связью между числом тринуклеотидных повторов и тяжестью клинических проявлений заболевания с цитологической экспрессией ломкости Х-хромосомы.
Таблица 6.3. Некоторые заболевания человека,
связанные с экспансией тринуклеотидных повторов
Вид патологии |
Локализация |
Тринуклеотидный |
Число тринуклеотидных |
|
|
|
гена |
повтор |
повторов |
|
|
|
|
|
|
|
|
|
|
|
норма |
патология |
|
Синдром ломкой |
Xq 27,3 |
ЦГГ |
5—50 |
>200 |
|
Х-хромосомы (FRAXA) |
|
|
|
|
|
Спино-бульбарная |
Xq 11-12 |
ЦАГ |
17—26 |
40—52 |
|
мышечная атрофия |
|
|
|
|
|
Миотоническая |
19q 13,3 |
ЦТГ |
5—27 |
50—1600 |
|
дистрофия |
|
|
|
|
274 |
|
|
|
|
|
|

Хорея Гентинггона |
4р 16,3 |
ЦАГ |
11—34 |
>42 |
Увеличение числа тринуклеотидных повторов и связанное с этим явление антиципации обнаружены при целом ряде заболеваний (табл. 6.3). Например, при аутосомно-доминантном заболевании—хорее Гетинггона выявляется четкая корреляция между числом ЦАГ-повторов и возрастом дебюта заболевания. У потомков пораженных отцов обнаруживается более тяжелое клиническое течение заболевания. Экспансия числа тринуклеотидных повторов происходит в мужском гаметогенезе.
Таким образом, в настоящее время описан новый класс наследственных болезней (около 10 заболеваний), при которых проявляется феномен антиципации, материнский или отцовский эффект, варьирующая пенетрантность. Установлена связь указанных особенностей наследования и фенотипического проявления этих заболеваний с возникновением динамических мутаций, приводящих к экспансии тринуклеотидных повторов.
Генетические болезни соматических клеток частично описаны в разд. 4.2.1.
6.4.2. Особенности человека как объекта генетических исследований
Основные закономерности наследственности и изменчивости живых организмов были открыты благодаря разработке и применению гибридологического метода генетического анализа, основоположником которого является Г. Мендель. Наиболее удобными объектами, широко используемыми генетиками для гибридизации и последующего анализа потомства, стали горох, дрозофила, дрожжи, некоторые бактерии и другие виды, легко скрещивающиеся в искусственных условиях. Отличительной особенностью этих видов является достаточно высокая плодовитость, позволяющая применять статистический подход при анализе потомства. Короткий жизненный цикл и быстрая смена поколений позволяют исследователям в относительно небольшие промежутки времени наблюдать передачу признаков в последовательном ряду многих поколений. Немаловажной характеристикой видов, используемых в генетических экспериментах, является также небольшое число групп сцепления в их геномах и умеренное модифицирование признаков под влиянием окружающей среды.
С точки зрения приведенных выше характеристик видов, удобных для применения гибридологического метода генетического анализа, человек как вид обладает целым рядом особенностей, не позволяющих применять этот метод для изучения его наследственности и изменчивости. Во-первых, у человека не может быть произведено искусственного направленного скрещивания в интересах исследователя. Во-вторых, низкая плодовитость делает невозможным применение статистического подхода при оценке немногочисленного потомства одной пары родителей. В-третьих, редкая смена поколений, происходящая в среднем через 25 лет, при значительной продолжительности жизни дает возможность одному исследователю наблюдать не более 3—4 последовательных поколений. Наконец,
275
medwedi.ru
изучение генетики человека затрудняется наличием в его геноме большого числа групп сцепления генов (23 у женщин и 24 у мужчин), а также высокой степенью фенотипического полиморфизма, связанного с влиянием среды.
Все перечисленные особенности человека делают невозможным применение для изучения его наследственности и изменчивости классического гибридологического метода генетического анализа, с помощью которого были открыты все основные закономерности наследования признаков и установлены законы наследственности. Однако генетиками разработаны приемы, позволяющие изучать наследование и изменчивость признаков у человека, несмотря на перечисленные выше ограничения.
Невозможность направленного скрещивания, проводимого в интересах исследования, и малочисленность потомства, получаемого от каждой родительской пары, компенсируются подбором в популяции семей с интересующим генетика признаком в количестве, достаточном для проведения статистического анализа потомства. Ограниченность числа поколений, которые может наблюдать один генетик, компенсируется возможностью подбора и регистрации последовательных поколений семей с интересующим признаком многими поколениями исследователей. Существенно облегчается генетический анализ у человека благодаря высокой степени изученности его фенотипа методами морфологии, физиологии, биохимии, иммунологии, клиники. Большие перспективы в изучении наследственности и изменчивости у человека открываются в связи с применением ранее используемых и новых методов генетических исследований.
6.4.3.Методы изучения генетики человека
Кметодам, широко используемым при изучении генетики человека, относятся генеалогический, популяционно-статистический, близнецовый, метод дерматоглифики, цитогенетический, биохимический, методы генетики соматических клеток.
6.4.3.1.Генеалогический метод
В основе этого метода лежит составление и анализ родословных. Этот метод широко применяют с древних времен и до наших дней в коневодстве, селекции ценных линий крупного рогатого скота и свиней, при получении чистопородных собак, а также при выведении новых пород пушных животных. Родословные человека составлялись на протяжении многих столетий в отношении царствующих семейств в Европе и Азии.
Как метод изучения генетики человека генеалогический метод стали применять только с начала XX столетия, когда выяснилось, что анализ родословных, в которых прослеживается передача из поколения в поколение какогото признака (заболевания), может заменить собой фактически неприменимый в отношении человека гибридологический метод.
276
При составлении родословных исходным является человек — пробанд, родословную которого изучают. Обычно это или больной, или носитель определенного признака, наследование которого необходимо изучить. При составлении родословных таблиц используют условные обозначения, предложенные Г. Юстом в 1931 г. (рис. 6.24). Поколения обозначают римскими цифрами, индивидов в данном поколении — арабскими.
Рис. 6.24. Условные обозначения при составлении родословных (по Г. Юсту)
С помощью генеалогического метода может быть установлена наследственная обусловленность изучаемого признака, а также тип его наследования (аутосомнодоминантный, аутосомно-рецессивный, X-сцепленный доминантный или рецессивный, Y-сцепленный). При анализе родословных по нескольким признакам может быть выявлен сцепленный характер их наследования, что используют при составлении хромосомных карт. Этот метод позволяет изучать интенсивность мутационного процесса, оценить экспрессивность и пенетрантность аллеля. Он широко используется в медико-генетическом консультировании для прогнозирования потомства. Однако необходимо отметить, что генеалогический анализ существенно осложняется при малодетности семей.
Родословные при аутосомно-доминантном наследовании. Для аутосомного типа наследования в целом характерна равная вероятность встречаемости данного признака как у мужчин, так и у женщин. Это обусловлено одинаковой двойной дозой генов, расположенных в аутосомах у всех представителей вида и получаемых от обоих родителей, и зависимостью развивающегося признака от характера взаимодействия аллельных генов.
При доминировании признака в потомстве родительской пары, где хотя бы
277
medwedi.ru
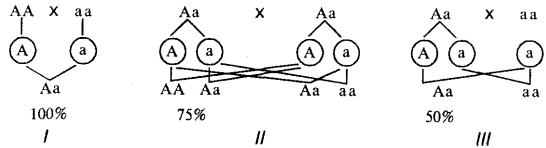
один родитель является его носителем, он проявляется с большей или меньшей вероятностью в зависимости от генетической конституции родителей (рис. 6.25).
Рис. 6.25. Вероятность появления потомков с доминантным признаком от различных супружеских пар (/—III)
Если анализируется признак, не влияющий на жизнеспособность организма, то носители доминантного признака могут быть как гомо-, так и гетерозиготами. В случае доминантного наследования какого-то патологического признака (заболевания) гомозиготы, как правило, нежизнеспособны, а носители этого признака — гетерозиготы.
Таким образом, при аутосомно-доминантном наследовании признак может встречаться в равной мере у мужчин и у женщин и прослеживается при достаточном по численности потомстве в каждом поколении по вертикали. Анализируя родословные, необходимо помнить о возможности неполного пенетрирования доминантного аллеля, обусловленной взаимодействием генов или факторами среды. Показатель пенетрантности может быть вычислен как отношение фактического числа носителей признака к числу ожидаемых носителей этого признака в данной семье. Необходимо также помнить, что некоторые заболевания проявляются не сразу с момента рождения ребенка. Многие болезни, наследуемые по доминантному типу, развиваются лишь в определенном возрасте. Так, хорея Гентингтона клинически проявляется к 35—40 годам, поздно проявляется и поликистоз почек. Поэтому при прогнозировании подобных заболеваний в расчет не принимаются братья и сестры, не достигшие критического возраста.
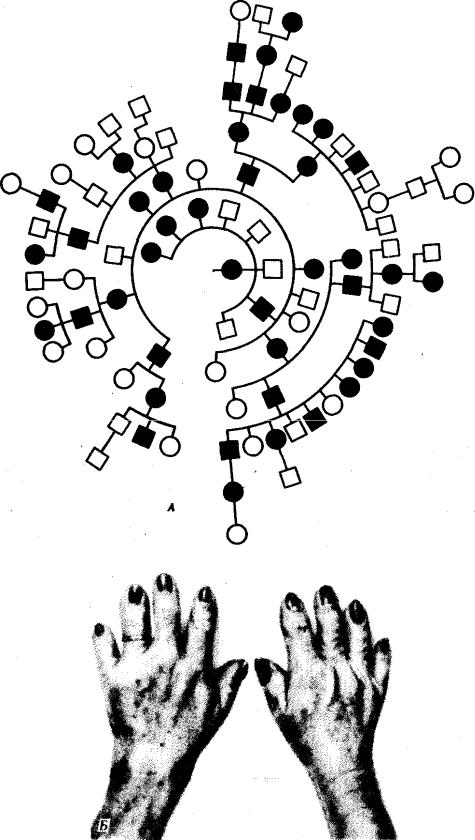
Первое описание родословной с аутосомно-доминантным типом наследования аномалии у человека было дано в 1905 г. В ней прослеживается передача в ряду поколений брахидактилии (короткопалости). На рис. 6.26 приведена родословная с этой аномалией. На рис. 6.27 изображена родословная с ретинобластомой в случае неполной пенетрантности.
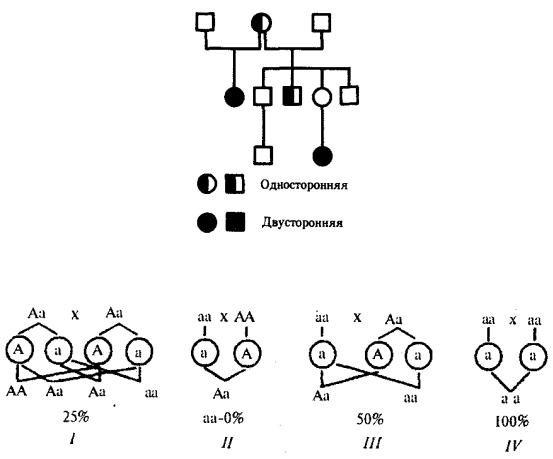
Родословные при аутосомно-рецессивном наследовании. Рецессивные признаки проявляются фенотипически лишь у гомозигот по рецессивным аллелям. Эти признаки, как правило, обнаруживаются у потомков фенотипически нормальных родителей — носителей рецессивных аллелей. Вероятность появления рецессивного потомства в этом случае равна 25%. Если один из родителей имеет рецессивный признак, то вероятность проявления его в потомстве будет зависеть от генотипа другого родителя. У рецессивных родителей все потомство унаследует
278
соответствующий рецессивный признак (рис. 6.28).
Рис. 6.26. Родословная (А) при аутосомно-доминантном типе наследования
279
medwedi.ru
(брахидактилия — Б)
Для родословных при аутосомно-рецессивном типе наследования характерно, что признак проявляется далеко не в каждом поколении. Чаще всего рецессивное потомство появляется у родителей с доминантным признаком, причем вероятность появления такого потомства возрастает в близкородственных браках, где оба родителя могут являться носителями одного и того же рецессивного аллеля, полученного от общего предка. Примером аутосомно-рецессивного наследования является родословная семьи с псевдогипертрофической прогрессивной миопатией, в
которой часты близкородственные браки (рис. 6.29). Обращает внимание распространение заболевания в последнем поколении по горизонтали.
Рис. 6.27. Родословная с ретинобластомой в случае неполной пенетрантности
Рис. 6.28. Вероятность появления потомков с рецессивным признаком от различных супружеских пар (I—IV)
Родословные при доминантном Х-сцепленном наследовании признака.
Гены, расположенные в Х-хромосоме и не имеющие аллелей в Y-хромосоме, представлены в генотипах мужчин и женщин в разных дозах. Женщина получает две свои Х-хромосомы и соответствующие гены как от отца, так и от матери, а мужчина наследует свою единственную Х-хромосому только от матери. Развитие соответствующего признака у мужчин определяется единственным аллелем, присутствующим в его генотипе, а у женщин он является результатом
280