

31-60
.docxАммиак – очень токсичное вещество, особенно для нервной системы. При физиологических значениях рН молекула NН3 легко превращается в ион аммония NН4+, который не способен проникать через биологические мембраны и задерживается в клетке. Накопление NН4+ вызывает торможение заключительных этапов цикла трикарбоновых кислот и снижение продукции АТФ.

Аммиак, образующийся в тканях, сначала превращается в нетоксичное соединение и в таком виде переносится кровью к печени или почкам. Такими транспортными формами являются аминокислоты глутамин, аспарагин и аланин.
Образование глутамина и аспарагина из глутамата и аспартата соответственно происходит во многих тканях, включая головной мозг:
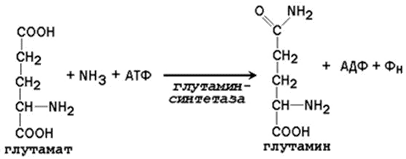

Глутамин - нейтральное нетоксичное соединение, способное легко проходить через клеточные мембраны. В виде этой аминокислоты аммиак транспортируется в крови. В крови здоровых людей содержание глутамина существенно превышает содержание других аминокислот. Глутамин, помимо участия в синтезе белка, служит источником азота в биосинтезе гистидина, глюкозамина, пуриновых и пиримидиновых нуклеотидов. С кровью глутамин поступает в печень и почки. Здесь он под действием фермента глутаминазы превращается в глутамат и аммиак. При участии аспарагиназы также происходит образование аммиака из аспарагина.
Аланин является транспортной формой аммиака, которая образуется преимущественно в мышцах. При интенсивной физической нагрузке источниками аммиака служат реакции дезаминирования аминокислот и аденозинмонофосфата (АМФ). Сначала аммиак превращается в аминогруппу глутамата в реакции восстановительного аминирования , катализируемой глутаматдегидрогеназой:

Образовавшийся глутамат переносит затем свою α-аминогруппу на пируват, всегда имеющийся в достаточном количестве, поскольку это продукт протекающего в мышцах гликолиза. Реакция катализируется аланинаминотрансферазой.
Глутамат + Пируват α-Кетоглутарат + Аланин
Аланин (нейтральная аминокислота, не несущая суммарного заряда при значениях рН, близких к 7) выходит из клеток и доставляется кровью к печени. Здесь он под действием аланинаминотрансферазы передаёт свою аминогруппу α-кетоглутарату, в результате чего образуется глутамат.
α-Кетоглутарат + Аланин Глутамат + Пируват
Далее в реакции, катализируемой глутаматдегидрогеназой, глутамат дезаминируется с образованием α-кетоглутарата и аммиака, который в печени превращается в мочевину.
39.
Биосинтез мочевины – основной путь обезвреживания аммиака. Мочевина синтезируется в орнитиновом цикле, протекающем в клетках печени. Эту последовательность реакций открыли Х.Кребс и К.Хензелейт в 1932 г. Согласно современным представлениям, цикл мочевины включает последовательность пяти реакций.
Две начальные реакции биосинтеза мочевины происходят в митохондриях клеток печени.


Последующие реакции протекают в цитоплазме клеток печени.




Схема орнитинового цикла и его связь с превращениями фумаровой и аспарагиновой кислот.
Цифрами обозначены ферменты, катализирующие реакции орнитинового цикла: 1 – карбамоилфосфатсинтетаза; 2 – орнитин-карбамоилтрансфераза; 3 – аргининосукцинатсинтетаза; 4 – аргининосукцинатлиаза; 5 – аргиназа.
Орнитиновый цикл находится в тесной взаимосвязи с циклом трикарбоновых кислот:
-пусковые реакции цикла мочевины, как и реакции ЦТК, протекают в митохондриальном матриксе;
-поступление СО2 и АТФ, необходимых для образования мочевины, обеспечивается работой ЦТК;
-в цикле мочевины образуется фумарат, который является одним из субстратов ЦТК. Фумарат гидратируется в малат, который в свою очередь окисляется в оксалоацетат. Оксалоацетат может подвергаться трансаминированию в аспартат; эта аминокислота участвует в образовании аргининосукцината.
Регуляция активности ферментов цикла осуществляется главным образом на уровне карбамоилфосфатсинтетазы, которая малоактивна в отсутствие своего аллостерического активатора - N-ацетил-глутамата. Концентрация последнего зависит от концентрации его предшественников (ацетил-КоА и глутамата), а также аргинина, который является аллостерическим активатором N-ацетилглутаматсинтазы:
Ацетил-КоА + Глутамат N-ацетилглутамат + КоА-SH
Концентрация ферментов орнитинового цикла зависит от содержания белка в пищевом рационе. При переходе на диету, богатую белком, в печени повышается синтез ферментов орнитинового цикла. При возвращении к сбалансированному рациону концентрация ферментов снижается. В условиях голодания, когда усиливается распад тканевых белков и использование аминокислот как энергетических субстратов, возрастает продукция аммиака, концентрация ферментов орнитинового цикла увеличивается.
Нарушения орнитинового цикла. Известны метаболические нарушения, обусловленные частичным блокированием каждого из 5 ферментов, катализирующих в печени реакции синтеза мочевины, а также N-ацетилглутаматсинтазы. Эти генетические дефекты, очевидно, являются частичными. Полное блокирование какой-либо из стадий цикла мочевины в печени, по-видимому, несовместимо с жизнью, потому что другого эффективного пути удаления аммиака не существует.
Общим признаком всех нарушений синтеза мочевины является повышенное содержание NH4+ в крови (гипераммониемия). Наиболее тяжёлые клинические проявления наблюдаются при дефекте фермента карбамоилфосфатсинтетазы. Клиническими симптомами, общими для всех нарушений цикла мочевины, являются рвота, нарушение координации движений, раздражительность, сонливость и умственная отсталость. Если заболевание не диагностируется, то быстро наступает гибель. У детей старшего возраста проявлениями заболевания служат повышенная возбудимость, увеличение размеров печени и отвращение к пище с высоким содержанием белка.
Лабораторная диагностика заболеваний включает определение содержания аммиака и метаболитов орнитинового цикла в крови, моче и спинномозговой жидкости; в сложных случаях прибегают к биопсии печени.
Значительное улучшение наблюдается при ограничении белка в диете, при этом могут быть предотвращены многие нарушения мозговой деятельности. Малобелковая диета приводит к снижению содержания аммиака в крови и к улучшению клинической картины при мягких формах этих наследственных нарушений. Пищу следует принимать часто, небольшими порциями, для того чтобы избежать резкого повышения уровня аммиака в крови.
Клинико-диагностическое значение определения мочевины в крови и моче. В крови здорового человека содержание мочевины составляет 3,33 – 8,32 ммоль/л. За сутки с мочой выводится 20 – 35 г мочевины.
Изменения содержания мочевины в крови при заболеваниях зависят от соотношения процессов её образования в печени и выведения почками. Повышение содержания мочевины в крови (гиперазотемия) отмечается при почечной недостаточности, снижение – при недостаточности печени, при диете с низким содержанием белков.
Повышение экскреции мочевины с мочой наблюдается при употреблении пищи с высоким содержанием белков, при заболеваниях, сопровождающихся усилением катаболизма белков в тканях, при приёме некоторых лекарств (например, салицилатов). Снижение экскреции мочевины с мочой характерно для заболеваний и токсических поражений печени, заболеваний почек, сопровождающихся нарушением их фильтрационной способности.
40.


41.
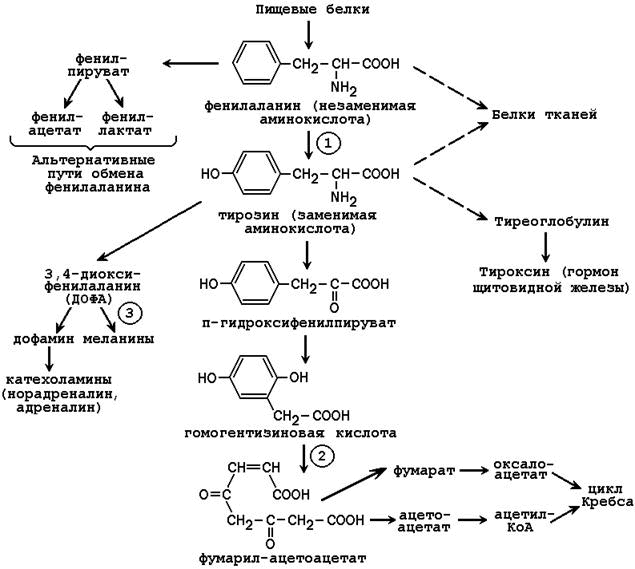
Известен ряд врождённых нарушений обмена фенилаланина и тирозина.
Фенилкетонурия – врождённое нарушение процесса гидроксилирования фенилаланина до тирозина. Заболевание чаще всего вызвано отсутствием или недостатком фермента фенилаланингидроксилазы (обозначен цифрой 1 на рисунке 25.2), реже - нарушением образования тетрагидробиоптерина.
Ранними симптомами фенилкетонурии являются повышенная возбудимость и двигательная активность, рвота и трудности вскармливания, с 3 – 5-го месяца нарушается интеллектуальное развитие, исчезает реакция на окружающее. Со временем у детей появляются судороги. Волосы и глаза обычно менее пигментированы, чем у других членов семьи. При отсутствии лечения продолжительность жизни больных составляет 20 - 30 лет.
Биохимическая основа фенилкетонурии – накопление фенилаланина в организме. Высокая концентрация аминокислоты стимулирует выработку фермента, превращающего фенилаланин в фенилпируват (в норме этот фермент малоактивен). Путём восстановления фенилпируват переходит в фениллактат, а путём декарбоксилирования – в фенилацетат. Эти продукты наряду с фенилаланином в существенных количествах обнаруживаются в моче больных.
В настоящее время имеются достоверные свидетельства того, что за токсическое повреждение мозга ответственны главным образом высокие концентрации фенилаланина. Повышенное содержание фенилаланина тормозит транспорт тирозина и других аминокислот через биологические мембраны. Это приводит к ограничению синтеза белка в клетках мозга и нарушению синтеза нейромедиаторов.
Раннюю диагностику заболевания нельзя провести исходя только из клинической симптоматики. Диагноз ставится биохимически путём скрининга всех новорождённых. Лечение больных фенилкетонурией основано на ограничении поступления фенилаланина в организм и снижения концентрации этой аминокислоты в плазме. С этой целью используются искусственные питательные смеси, в которых фенилаланин отсутствует (например, берлофен).
Алкаптонурия – врожденное нарушение обмена фенилаланина, вызванное отсутствием фермента оксидазы гомогентизиновой кислоты (цифра 2 на рисунке 25.2). Это приводит к нарушению образования малеилацетоацетата, расщепляющегося далее до фумарата и ацетоацетата. В раннем детском возрасте единственным проявлением дефицита фермента является изменение окраски мочи. Гомогентизиновая кислота секретируется в просвет канальцев и в значительном количестве выводится с мочой. На воздухе она окисляется, а затем полимеризуется в окрашенное соединение, которое окрашивает пелёнки в чёрный цвет. Экскреция гомогентизиновой кислоты зависит от содержания фенилаланина и тирозина в пище.
Следствием накопления гомогентизиновой кислоты в организме является охроноз - шиферно-голубой оттенок ушного и носового хрящей, вызванный накоплением в них пигмента. Развитие охроноза можно предотвратить, если с раннего возраста ограничивать поступление с пищей фенилаланина и тирозина.
Альбинизм развивается при отсутствии в пигментных клетках фермента тирозиназы (обозначена цифрой 3 на рисунке 25.2), которая участвует в образовании меланина. В результате волосы, кожа и глаза больного лишены этого пигмента. При альбинизме наблюдается повышение чувствительности к солнечным лучам и некоторое нарушения зрения.
42.
Метильная группа метионина, связанная с атомом серы, также представляет собой подвижную одноуглеродную группу, способную участвовать в реакциях трансметилирования (переноса метильной группы). Активной формой метионина, принимающей непосредственное участие в этих превращениях, является S-аденозилметионин, который образуется при взаимодействии метионина с АТФ.
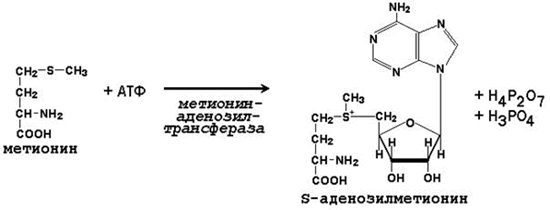
Примеры реакций трансметилирования с участием S-аденозилметионина(Субстрат-Метилированный продукт)
Норадреналин-Адреналин
Адреналин-Метоксиадреналин
Гуанидинацетат-Креатин
Карнозин-Ансерин
Гистамин-N-метилгистамин
Фосфатидилэтаноламин-Фосфатидилхолин
После отдачи метильной группы S-аденозилметионин превращается в S-аденозилгомоцистеин. Последний расщепляется на аденозин и гомоцистеин. Гомоцистеин может вновь превращаться в метионин за счёт метильной группы 5-метил-ТГФК
 (дальше
верхняя реакция)
(дальше
верхняя реакция)
В этой реакции в качестве кофермента участвует метилкобаламин – производное витамина В12. При недостатке витамина В12 нарушается синтез метионина из гомоцистеина и накапливается 5-метил-ТГФК. Так как реакция образования 5-метил-ТГФК из 5,10-метилен-ТГФК необратима, одновременно возникает дефицит фолиевой кислоты.
43.
Главную роль в реакциях обмена серина и глицина играют ферменты, в состав которых в качестве кофермента входит тетрагидрофолиевая кислота (ТГФК). ТГФК образуется в организме в результате восстановления фолиевой кислоты (витамина Вс).

фолиевая кислота
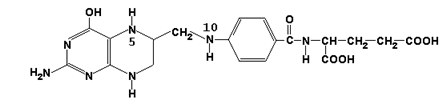
ТГФК
Реакционноспособными центрами в молекуле ТГФК являются атомы азота в положениях 5 и 10. Атомы водорода при N5 и N10 могут замещаться на различные одноуглеродные группы: метильную (-СН3), метиленовую (-СН2-), метенильную (=СН-), формильную (-СН=О) и некоторые другие. Основными источниками одноуглеродных групп в клетке служат серин и глицин.
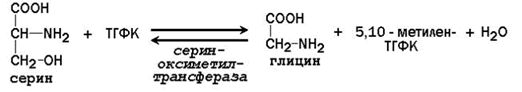

5,10-Метилен-ТГФК используется как донор метильной группы в реакциях биосинтеза тимидилового нуклеотида.
При окислении 5,10-метилен-ТГФК образуются 5,10-метенил-ТГФК и 10-формил-ТГФК. Эти производные ТГФК служат источниками атомов углерода в процессе биосинтеза пуриновых нуклеотидов (аденилового и гуанилового).
При восстановлении 5,10-метилен-ТГФК образуется 5-метил-ТГФК. Это соединение интересно тем, что может поставлять метильную группу для регенерации метионина из гомоцистеина (см. далее).
Аминокислота глицин, помимо участия в синтезе белка и образовании различных одноуглеродных групп, является предшественником ряда специализированных биомолекул:
-оба атома углерода и атом азота глицина могут включаться в структуру пуринового ядра (атомы С4, С5 и N7);
-глицин является главным предшественником порфиринов (простетической группы гемоглобина, миоглобина, цитохромов);
-глицин участвует в синтезе креатина - предшественника креатинфосфата, участвующего в биоэнергетике мышечной и нервной ткани;
-глицин входит в состав пептидного кофермента глутатиона;
-участвует в образовании конъюгатов (гликохолевая кислота, гиппуровая кислота).
44.
Схема биосинтеза гемоглобина представлена на рисунке 24.1. Исходными веществами в этом метаболическом пути являются аминокислота глицин и метаболит цикла Кребса сукцинил-КоА. Синтез происходит в ретикулоцитах (незрелых эритроцитах, содержащих клеточное ядро). Реакции идут в митохондриях и цитоплазме клеток.
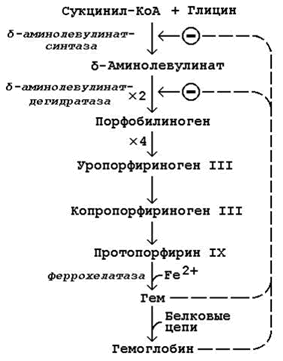
Биосинтез гемоглобина и его регуляция.
Первая стадия в последовательности реакций, ведущих к синтезу гема, катализируется δ-аминолевулинат-синтазой. Фермент абсолютно специфичен к субстратам; кофакторами фермента являются пиридоксаль-5-фосфат и ионы Mg2+.

Имеются данные о том, что некоторые лекарственные препараты, а также стероидные гормоны, напротив, индуцируют синтез печёночной δ-аминолевулинат-синтазы.
Во второй реакции, катализируемой δ-аминолевулинат-дегидратазой, при конденсации двух молекул δ-аминолевулината образуется порфобилиноген.

В дальнейшем из четырёх молекул порфобилиногена в результате ряда сложных ферментативных реакций образуется протопорфирин IX – непосредственный предшественник гема. При участии митохондриального фермента феррохелатазы двухвалентное железо включается в уже готовую структуру протопорфирина. Для протекания этой реакции необходимы аскорбиновая кислота и цистеин в качестве восстановителей. Ингибитором феррохелатазы является свинец. На заключительном этапе происходит соединение гема с белковыми цепями, характерными для синтезируемого хромопротеина. Конечные продукты этого биосинтеза (гем, гемоглобин) подавляют начальные реакции по механизму отрицательной обратной связи.
При врождённых и приобретённых нарушениях биосинтеза гема развиваются заболевания – порфирии.
Порфирии – группа наследственных заболеваний, обусловленных частичным дефицитом одного из ферментов синтеза гема. Снижение образования гема приводит к снятию его ингибирующего эффекта на начальные этапы биосинтеза, результатом чего является избыточное образование порфиринов и их предшественников. Основными симптомами порфирий являются:
-нарушения со стороны центральной нервной системы (т.к. предшественники порфиринов являются нейротоксинами);
-повышенная светочувствительность кожи (порфирины накапливаются в коже, поглощают свет и переходят в возбуждённое состояние, вызывая образование токсичных свободных радикалов);
-анемия (снижение содержания гемоглобина в крови) ;
-порфиринурия - выведение порфиринов с мочой и калом (моча приобретает красную окраску).
Порфиринурия может также развиваться при отравлениях свинцом.
Содержание гемоглобина в крови здоровых людей составляет 130-160 г/л. Гемоглобин крови полностью обновляется в течение 120 дней (продолжительность жизни эритроцита).
Разрушение эритроцитов и начальные этапы катаболизма гема происходят в клетках ретикуло-эндотелиальной системы (РЭС), которые находятся в печени (клетки Купфера), селезёнке, костном мозге.
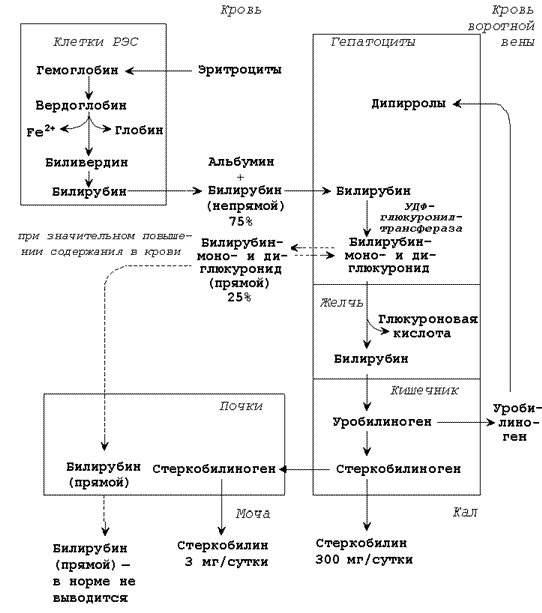
Схема катаболизма гемоглобина в тканях.
Продукты распада гема называют желчными пигментами, так как все они в разных количествах обнаруживаются в желчи. К желчным пигментам относятся: биливердин (зелёного цвета), билирубин (красно-коричневого цвета), уробилиноген и стеркобилиноген (бесцветные), уробилин и стеркобилин (жёлтого цвета). Далее приводятся формулы билирубина и его диглюкуронида. Билирубин (свободный или неконъюгированный билирубин) образуется в клетках ретикуло-эндотелиальной системы (РЭС), транспортируется в гепатоциты. Билирубин нерастворим в воде и растворим в жирах, токсичен, в крови присутствует в виде комплекса с альбумином, не проникает через почечный фильтр.
Эта фракция билирубина в плазме крови называется непрямым билирубином, так как взаимодействует с диазореактивом только после осаждения альбуминов.

Билирубиндиглюкуронид (связанный или конъюгированный билирубин) образуется в гепатоцитах под действием фермента билирубин-глюкуронилтрансферазы, путём активного транспорта выводится в желчные канальцы. Он хорошо растворим в воде и не растворим в жирах, малотоксичен, в крови не связан с белками плазмы, может проникать через почечный фильтр. Эта фракция билирубина в плазме крови называется прямым билирубином, так как непосредственно может взаимодействовать с диазореактивом.

Общее содержание билирубина в крови здорового человека составляет 8 – 20 мкмоль/л, из них 6 – 15 мкмоль/л приходится на непрямой билирубин, 2 – 5 мкмоль/л – на прямой билирубин. Увеличение общего билирубина в крови (более 27 мкмоль/л) приводит к окрашиванию кожи, слизистых оболочек, склеры глаз в жёлтый цвет (желтуха). Определение содержания желчных пигментов в крови используют при выяснении происхождения желтух. Желтуха бывает надпечёночная (гемолитическая), печёночная (паренхиматозная), подпечёночная (обтурационная или механическая).
Надпечёночная (гемолитическая) желтуха вызвана массивным распадом эритроцитов в результате резус-конфликта, попадания в кровь веществ, вызывающих разрушение мембран эритроцитов и некоторых других заболеваниях. При этой форме желтухи в крови повышено содержание непрямого билирубина, в моче повышено содержание стеркобилина, билирубин отсутствует, в кале повышено содержание стеркобилина.
Печёночная (паренхиматозная) желтуха вызвана повреждением клеток печени при инфекциях и интоксикациях. При этой форме желтухи в крови повышено содержание непрямого и прямого билирубина, в моче повышено содержание уробилина, присутствует билирубин, в кале понижено содержание стеркобилина.
Подпечёночная (обтурационная) желтуха вызвана нарушением оттока желчи, например, при закупорке желчевыводящего протока камнем. При этой форме желтухи в крови повышено содержание прямого билирубина (иногда и непрямого), в моче отсутствует стеркобилин, присутствует билирубин, в кале понижено содержание стеркобилина.
Условно физиологическая желтуха новорождённых развивается у большинства здоровых новорождённых в первые дни после рождения и продолжается около двух недель. При различных заболеваниях, возникающих у новорождённых, а также у недоношенных детей желтушный период затягивается. Увеличение длительности гипербилирубинемии может привести к серьёзным последствиям: накоплению билирубина в ткани мозга (ядерная желтуха).
Повышению содержания билирубина в крови новорождённых могут способствовать следующие особенности обмена веществ в их организме:
-замена фетального гемоглобина на гемоглобин А. В первые дни после рождения усиливается гемолиз эритроцитов, содержващих HbF; образуются новые эритроциты, содержащие HbA. HbF подвергается катаболизму; образуется значительное количество билирубина;
-отвлечение альбуминов плазмы для транспорта жирных кислот. Содержание углеводов в организме новорождённых сравнительно невелико; основным энергетическим субстратом являются жирные кислоты, концентрация которых в крови повышается, жирные кислоты транспортируются в комплексе с альбуминами;
-низкая активность глюкуронилтрансферазы в ткани печени. Замедление процессов конъюгации билирубина в печени затрудняет его выведение в кишечник;
-стерильность кишечника. В кишечнике новорождённого отсутствует микрофлора, поэтому билирубин не превращается в стеркобилиноген и может происходить его обратное всасывание в кровоток.
45.
Нуклеиновыми кислотами или полинуклеотидами называются высокомолекулярные вещества, состоящие из нуклеотидов, соединённых в цепь 3', 5'-фосфодиэфирными связями. Каждый нуклеотид состоит из азотистого основания, углевода (пентозы) и остатка фосфорной кислоты.
Азотистые основания, входящие в состав нуклеотидов, имеют следующее строение:

Углеводы представлены рибозой и дезоксирибозой:

Азотистое основание и пентоза, соединённые N-гликозидной связью, образуют нуклеозид. Если в качестве пентозы в нуклеозиде присутствует рибоза, то это рибонуклеозид, а если дезоксирибоза - то это дезоксирибонуклеозид. Примеры нуклеозидов:

Нуклеотиды представляют собой фосфорилированные нуклеозиды. Остаток фосфорной кислоты, как правило, присоединяется к гидроксильной группе пентозы в 5'-положении при помощи сложноэфирной связи. Примеры:

В клетках встречаются также нуклеозиддифосфаты и нуклеозидтрифосфаты, содержащие соответственно два и три остатка фосфорной кислоты. Биологическая роль этих соединений будет рассматриваться в дальнейшем.
Кроме перечисленных, известны минорные нуклеотиды (редко встречающиеся) . Они содержат, как правило, метилированные производные вышеприведённых главных азотистых оснований. Минорные основания присутствуют в составе некоторых разновидностей рибонуклеиновых кислот. Роль этих оснований заключается, очевидно, в защите молекулы нуклеиновой кислоты от действия гидролитических ферментов.
Входят в состав ДНК и РНК.
46.
Ключевым соединением в биосинтезе как пуриновых, так и пиримидиновых нуклеотидов является 5-фосфорибозил-1-пирофосфат (ФРПФ). Это соединение участвует также в синтезе коферментов НАД+ и НАДФ+.
ФРПФ образуется при взаимодействии рибозо-5-фосфата и АТФ. Источниками рибозофосфата служат пентозофосфатный путь и распад нуклеотидов. Катализирует реакцию фермент ФРПФ-синтаза.

Внутриклеточная концентрация ФРПФ обычно низкая и строго регулируется. Скорость синтеза ФРПФ зависит от наличия субстратов синтеза, особенно рибозо-5-фосфата, и каталитической активности ФРПФ-синтазы, на которую влияют концентрация неорганического фосфата и концентрация АМФ, ГМФ и ИМФ, выступающих в качестве эффекторов.
Молекула ФРПФ служит основой для последующего синтеза пуринового ядра. Источниками атомов углерода и азота являются аминокислоты глутамин, глицин и аспартат, СО2 и два одноуглеродных производных ТГФК – формил-ТГФК и метенил-ТГФК.

Происхождение атомов пуринового ядра.
Сначала в реакции, катализируемой фосфорибозил-пирофосфат-амидотрансферазой, из ФРПФ при участии глутамина образуется 5-фосфорибозиламин.

ФРПФ-амидотрансфераза – второй регуляторный фермент синтеза пуриновых нуклеотидов, он ингибируется АМФ и ГМФ по принципу обратной связи. Роль этого фермента в биосинтезе пуринов de novo , однако, менее существенна, чем ФРПФ-синтазы.