

Эволюция понятия «валентность» и его роль в истории химии
В начале 19 в. Дж. Дальтоном был сформулирован закон кратных отношений, из которого следовало, что каждый атом одного элемента может соединяться с одним, двумя, тремя и т.д. атомами другого элемента (как, например, в окислах азота —N2O, NO, N2O3, NO2 и N2O5). В середине 19 в., когда были определены точные относительные веса атомов (И. Я. Берцелиус и др.), стало ясно, что наибольшее число атомов, с которыми может соединяться данный атом, не превышает определённой величины, зависящей от его природы. Например, атом F может соединяться лишь с одним атомом Н, О — с двумя, N — с тремя, С — с четырьмя, образуя соответственно HF, H2O, NH3 и CH4. Два или четыре атома Н в метане CH4 могут быть замещены одним или двумя атомами О с образованием формальдегида CH2O и двуокиси углерода CO2 соответственно, три атома Н в CH4 могут замещаться одним атомом N с образованием цианистого водорода HCN, и т.д. Эта способность связывать или замещать определённое число других атомов и была названа «Валентность» (Э. Франкленд, 1853).
В таком определении валентность, естественно, всегда выражается целыми числами. Поскольку в то время для водорода не были известны соединения, где он был бы связан более чем с одним атомом любого другого элемента, атом Н был выбран в качестве стандарта, обладающего валентностью, равной 1. В «водородной» шкале кислород и сера имеют валентность, равную 2, азот и фосфор 3, углерод и кремний 4. Однако «водородной» шкалы оказалось недостаточно: в других соединениях, например в окислах, один и тот же элемент может реализовать валентности, которые не осуществляются в гидридах (существуют окислы P2O5, SO3 и Cl2O7, но неизвестны гидриды PH5, SH6 и ClH7). В качестве второго стандарта с валентностью, равной 2, был выбран кислород.
В конце 50-х гг. 19 в. А. С. Купер и А.Кекуле постулировали принцип постоянной четырёхвалентности углерода в органических соединениях. Представления о валентности составили важную часть теории химического строения А. М. Бутлерова (1861). Образование химической связи рассматривалось как результат взаимного насыщения двухвалентной пары взаимодействующих атомов (по одной валентности от каждого), кратные связи соответствовали насыщению нескольких валентностей от каждого атома, и т.д. Каждая связь считалась локализованной между двумя атомами и изображалась одной чертой, соединяющей эти атомы. Молекулы стали изображать с помощью структурных формул, получивших особенно широкое распространение в органической химии.
Положения Бутлерова в дальнейшем легли в основу структурной теории, рассматривающей и пространственное расположение атомов в молекуле. Было найдено, что простые молекулы типа MXk с одинаковым центральным атомом M и разными заместителями Х имеют схожее геометрическое строение. Независимость геометрического строения от типа связи в широких пределах привела к мысли, что пространственное расположение атомов в молекулах MXk определяется валентностью центрального атома М и что эти валентности имеют направленный характер.
Периодический закон Д. И. Менделеева (1869) вскрыл зависимость валентности элемента от его положения в периодической системе. Элементы одинаковых групп системы обладают одинаковой высшей валентностью, в большинстве случаев равной номеру той группы, в которой находится этот элемент; высшая валентность меняется на 1 при переходе от одной группы к соседним.
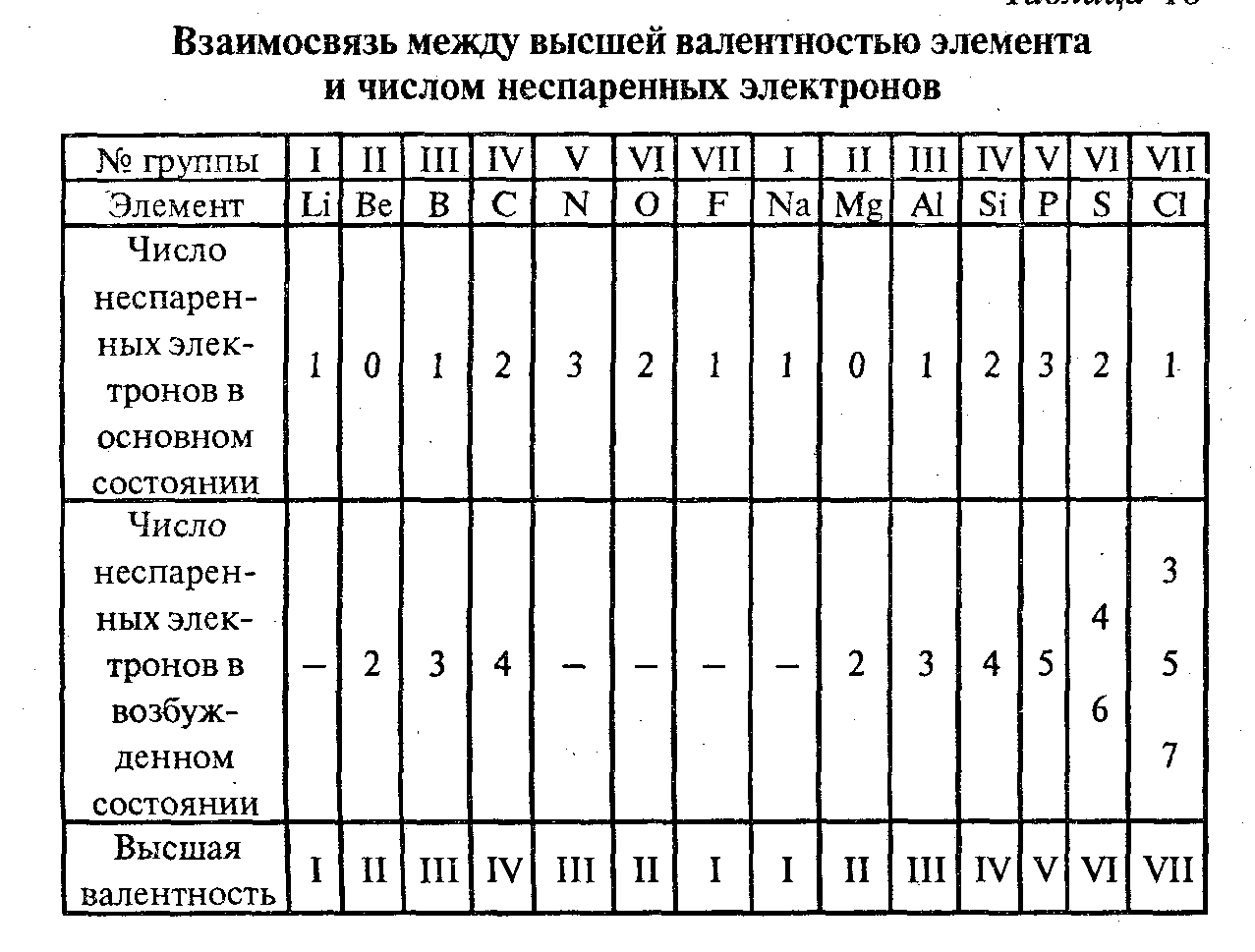
Эта зависимость сыграла чрезвычайно важную роль в развитии химии: зная лишь положение элемента (в том числе элементов, которые в то время ещё не были открыты) в периодической системе, можно было определить его валентные возможности, предсказать состав его соединений и впоследствии синтезировать их. С помощью представлений о формальной (стехиометрической) валентности химикам удалось обобщить и систематизировать огромный экспериментальный материал по строению, стехиометрическому составу и свойствам многих десятков и сотен тысяч органических и неорганических соединений.
Первые электронные теории ковалентности и гетеровалентности. До электронных представлений о строении вещества валентность трактовалась формально. Лишь в 20 в. было установлено, что химическая связь осуществляется за счёт электронов внешних (валентных) оболочек атомов.
В 1916 Г. Льюис постулировал, что химическая связь осуществляется парой электронов, принадлежащих одновременно обоим взаимодействующим атомам. В 1917 Коссель выдвинул гипотезу, согласно которой электронная пара связи переходит целиком к одному из атомов с образованием ионной пары катион — анион, удерживающихся в молекуле электростатическими силами. Согласно обеим гипотезам наиболее устойчивыми оказываются соединения, в которых валентные электроны распределялись так, чтобы каждый атом был окружен оболочкой, имитирующей электронную оболочку ближайшего инертного газа (правило октета). Гипотеза Льюиса положила начало электронной теории ковалентной связи и ковалентности, гипотеза Косселя — теории ионной связи и гетеровалентности. Обе представляли крайние случаи общей картины полярной связи, когда электронная пара смещена к одному из атомов лишь частично и степень смещения может варьироваться от 0 до 1. Валентность атома в соединении, согласно классической электронной теории, равна числу его неспаренных электронов, участвующих в связях, а максимальная валентность — обычно полному числу электронов в его валентной оболочке, то есть номеру группы периодической системы, в которой находится элемент. Элементы одинаковых групп имеют одинаковое число валентных электронов, а внутри одинаковых подгрупп — и одинаковые или очень близкие электронные конфигурации. Сходство строения валентных оболочек атомов обусловливает сходство их соединений.
Ковалентность и гетеровалентность отражают специфику соответствующего типа химической связи. Для ковалентности важна насыщаемость связей, обусловливающая существование молекул в виде дискретных частиц с определённым составом и структурой. Ковалентность эффективна для органических и большинства простых неорганических соединений. Напротив, в случае гетеровалентности максимальное число ионов противоположного знака, способное разместиться вокруг данного иона, в основном определяется соотношениями их размеров. Ионная валентность эффективна для сравнительно ограниченного класса соединений, в основном для различных солей, щелочных, щёлочноземельных и некоторых др. металлов.
Сейчас установлено, что подавляющее большинство элементов может проявлять переменную валентность, образуя весь ряд «валентноненасыщенных» соединений со всеми значениями валентности от 1 до максимальной с изменением на 1 (например, известны молекулы BF, BF2 и BF3; CF, CF2, CF3 и CF4 и т.д.). Валентность не может считаться жестко специфической характеристикой элемента, можно говорить лишь об относительной типичности или относительной устойчивости разных значений валентности. У непереходных элементов чётных и нечётных групп наиболее устойчивы соответственно чётные и нечётные валентности, например в молекулах типа PF3, PF5, SF2, SF4, SF6, IF, IF3, IF5, IF7 и т.д., где типичная валентность атомов Р, S и I изменяется на 2 единицы.
Радикалы типа ·PF4, ·SF3, ·SF5, ·IF2, ·IF4 и т.д. с четырёхвалентным фосфором, нечётновалентными аналогами серы и инертными газами и чётновалентными галогенами значительно менее стабильны, обладают отчётливо выраженной склонностью к отщеплению одного электрона (с образованием более устойчивых катионов типа PF4+, SF3+, SF5+, IF2+, IF4+) или одного атома заместителя и характеризуются значительно меньшими временами существования. У элементов побочных групп соотношения между типичными и менее типичными валентностями имеют более сложный характер.
Изучение электронных спектров показало, что двухатомные молекулы типа O2, S2, OS и др. имеют два неспаренных электрона; в рамках классических представлений это следовало бы интерпретировать так, будто в подобных молекулах каждый атом сохраняет неиспользованной одну свою валентность, хотя нет никаких видимых препятствий для их использования.
Таким образом, поиск общего определения валентности, охватывающего все известные типы соединений и тем более способного предсказать возможность или принципиальную невозможность существования ещё не известных классов соединений, представляет сложную проблему. Конечно, параллельно с «неклассическими» соединениями химиками были синтезированы многие сотни тысяч соединений, которые могут быть интерпретированы в рамках обычных классических представлений о валентности. Однако ясно, что все существующие частные определения валентности ограничены определёнными классами и типами соединений, в которых преобладает какой-либо один тип химического взаимодействия. В общем же случае связи имеют промежуточный характер между чисто ионными и чисто ковалентными, в них принимают участие все типы взаимодействия одновременно, но в различных количественных соотношениях, резко изменяющихся от класса к классу и более плавно — от соединения к соединению внутри одного класса. При отсутствии общего определения валентности трудность заключается в том, чтобы определить границы, где перестаёт быть справедливым одно частное определение валентности и его заменяет другое. Решить эту проблему только на основании экспериментальных фактов и классических представлений невозможно. Существенную помощь здесь может оказать квантовая теория химической связи и валентности.