

IV.Захворювання, які спричиняють деградацію розумового розвитку:
Хвороба Тея-Сакса (Амавротичне слабоумство)
Належить до групи внутрішньоклітинних ліпідозів. Це захворювання за утосомно-рецесивним типом успадкування. Відзначається збільшення в мозку гліколіпіду - гангліозиду, а також підвищення рівня гангліозидів у печінці, селезінці, що свідчить про генералізоване порушення обміну гангліозидів.
Гістологічно проявляється картина генералізованого розпаду гангліозних клітин нервової системи. Хвороба починається у віці 4-6 місяців. Часто це захворювання носить сімейний характер. У хворих рано виявляється зниження зору. Дитина не може фіксувати погляд, не стежить за іграшками. Досить рано на очному дні виявляється симптом "вишневої кісточки" - вишнево-червона цятка в макулярній ділянці, оточена сірувато-білим кільцем. Згодом розвиваються атрофія зорових нервів і повна сліпота. Зникають орієнтувальні і захисні реакції. Порушення призводять до повної нерухомості. При хворобі Тея - Сакса спостерігається симптом підвищеної реакції на звукові подразники - діти різко здригаються від звичайного звуку, можуть відзначатися судоми. Смерть настає в середньому через 1-2 роки після початку захворювання. Специфічне лікування амавротичної ідіотії не розроблено. Можливе встановлення гетерозиготного носія мутантного гена у батьків хворих. У випадку вагітності жінки, гетерозиготної за геном амавротичної ідіотії Тея - Сакса, доцільне дослідження гексоамінідази-А в амніотичній рідині, отриманій на 18-20-й тиж-день вагітності. При значному зниженні гексоамінідази-А показане штучне переривання вагітност
Синдром Пфаундлера-Гурлера (гаргоелізм)

Важке спадкове захворювання ; характеризується недостатністю альфа -L - ідуронідази - ферменту, що у катаболізме кислих мукополісахаридів, які складають основу міжклітинної речовини сполучної тканини. Виділяють дві алельних форми синдрому. Перша форма - власне синдром Гурлер, проявляється в перші місяці життя грубими рисами обличчя (гаргоілізм), запала переніссям, помутнінням рогівки, гепатоспленомегалією, тугоподвижністью суглобів, деформацією хребта; на 2 -му році життя з'являються коротка шия, широко розставлені очі, широкий ніс, товсті губи і язик, відкритий рот, маленькі рідкісні зуби; череп нерідко має форму кіля човна. Типові помутніння рогівки; тугоподвижность суглобів, деформації скелета; макроглоссія, гепатоспленомегалія; клапанні вади серця, аномалії коронарних артерій, ураження міокарда по типу гіпертрофічної кардіоміопатії; затримка росту . Психомоторний розвиток до 2 років, як правило, нормальне, далі відзначається виражена розумова відсталість, розвиваються глухота, сліпота. Прогноз несприятливий, хворі рідко доживають до 10 років. Друга форма - синдром Шейе, проявляється в шкільному віці незначним відставанням у рості, укороченням шиї і тулуба, тугоподвижністю суглобів, ураженням аортального клапана, помутнінням рогівки. Тече більш доброякісно, інтелект зазвичай не страждає. При обстеженні виявляють вакуолізіровані лімфоцити в мазках крові і підвищену екскрецію мукополісахаридів (дерматансульфат, гепарансульфат) з сечею. Тип спадкування - аутосомно -рецесивний . Можлива пренатальна діагностика за допомогою дослідження обміну мукополісахаридів в культурі клітин амніотичної рідини, одержуваної за допомогою трансабдомінального амніоцентеза. Лікування включає трансплантацію рогівки, застосування діуретиків та вазодилататорів при серцевій недостатності, антибактеріальну профілактику інфекційного ендокардиту при ураженнях клапанів серця, при необхідності - їх протезування.
Синдром Лоуренса-Муна-Бидля-Барді

рідкісна генетична патологія з аутосомно-рецесивним типом успадкування. Основними ознаками розладу є:
1 . Розумова відсталість (від глибокої до впритул наближається до кордонів норми);
2 . Пігментний ретиніт
3. Адипозогенитальная дистрофія. Можуть бути також полідактилія , синдактилія , деформація скелета і черепа , атрезія заднього проходу. Спостерігаються , крім того, ожиріння , гипогенитализм та епілептичні припадки. Зростання пацієнтів може бути збільшеним , зменшеним або нормальним . Розпізнавання хвороби можливо в дитячому віці за характерними дизгенетичними ознаками. Симптомокомплекс розлади стає чітко вираженим у віці до 1 -го року, чому нерідко сприяють менінгіти, енцефаліти, ЧМТ. Носійство рецесивного гена фенотипически ніяк себе не виявляє. Лікування симптоматичне, ефективних методів профілактики захворювання нині не існує.
Синдром Бурневіля
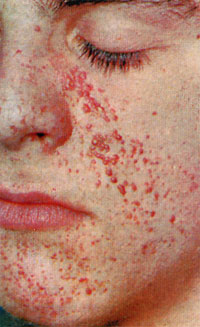
Гетерогенне, генетично детерміноване захворювання, що характеризується гіперплазією похідних екто-і мезодерми, ураженням шкіри, нервової системи і наявністю доброякісних пухлин (гамартом) в різних органах.
Клінічні симптоми хвороби Бурневілля-Прінгл з'являються в перші роки життя, але можуть існувати з народження. Процес поступово прогресує, особливо в період статевого дозрівання. Шкіра уражена в 96% випадків.