

Гель-эффект
Реакция обрыва является единственной элементарной реакцией, которая контролируется диффузией на всех стадиях процесса. С увеличением степени превращения мономера (конверсии), т.е. с его исчерпанием, скорость и степень полимеризации должны уменьшаться. Однако было обнаружено, что при полимеризации ММА увеличение степени превращения мономера приводит не к уменьшению, а к увеличению скорости и степени полимеризации. Это явление было названо гель-эффектом, поскольку оно наблюдалось при достаточно больших степенях превращения, когда вязкость реакционной среды заметно возрастала (примерно на два порядка по сравнению с первоначальной). Гель-эффект – это ускорение полимеризации на поздних стадияхвследствие увеличения времени жизни и концентрации макрорадикалов.
По мере протекания реакции полимеризации и возрастания вязкости среды возможность встречи макрорадикалов своими реакционно-способными группами становится все меньше и меньше. В результате падает скорость обрыва цепи и увеличивается время жизни радикалов, которое, например, при степени конверсии винилацетата, равной 80 %, достигает 30 с. Время жизни растущих радикалов мало (обычно несколько секунд). По мере роста макрорадикалов увеличивается вязкость системы, снижается подвижность макрорадикалов. В результате уменьшается скорость обрыва цепи путем рекомбинации. Время жизни радикалов возрастает также при снижении температуры. Рост времени жизни радикалов при увеличении вязкости системы приводит к интересному явлению ().
Длительная "жизнь" макрорадикала связана с низкой скоростью диффузии малоподвижных макрорадикалов в вязкой среде. При этом большая скорость диффузии мономерных молекул обеспечивает большое число встреч с макрорадикалами, (несмотря на малую подвижность последних) и рост цепи продолжается с высокой скоростью. Увеличение времени жизни макрорадикалов приводит к возрастанию их концентрации в реакционной смеси, а это, в свою очередь, к увеличению количества мономерных молекул, присоединившихся к ним в единицу времени, т.е. к увеличению общей скорости полимеризации. Одновременно растет степень полимеризации.
и большим значением предэкспоненциального множителя A в уравнении Аррениуса
|
|
|
|
|
|
отражающем малую вероятность столкновения макрорадикалов своими активными концами.
В соответствии с уравнением Смолуховского число встреч двух молекул жидкости равно
|
|
|
|
|
|
но так как
|
|
|
|
|
|
где KD – константа скорости встречи молекул; [A] и [B] – концентрации сталкивающихся частиц, R – расстояние между центрами тяжести частиц при встрече; D – сумма коэффициентов диффузии молекул A и B. Если подставить соответствующие значения, то для низкомолекулярных веществ KD 4109 л/мольс. Константа скорости таких веществ значительно меньше этой величины. Поэтому для низкомолекулярных веществ скорость реакции не лимитируется диффузиейДля макрорадикалов KD 107 л/мольс на малой глубине полимеризации. Эти значения соизмеримы с константой скорости реакции обрыва цепи. Поэтому дальнейшее снижение KD должно сильно отразиться на скорости рекомбинации. При глубоких степенях конверсии в реакционной системе начинаются существенные изменения ее состава и физических свойств, что отражается как на кинетике, так и на свойствах полимера. В первую очередь резко возрастает вязкость среды, что снижает скорость диффузии реагирующих частиц. Значительное увеличение вязкости реакционной среды ограничивает в первую очередь диффузионную подвижность макрорадикалов и, следовательно, снижает скорость обрыва, приводя к увеличению скорости радикальной полимеризации и молекулярной массы образующегося полимера (гель-эффект).
Неожиданное резкое ускорение полимеризации на второй стадии характерно для многих мономеров и носит название гель-эффект. ПприП При увеличении вязкости снижается движение больших макрорадикалов, а на передвижение маленьких молекул мономера вязкость влияет значительно меньше. Это приводит к увеличению средней продолжительности жизни кинетической цепи и увеличению концентрации макрорадикалов. Диффузия макрорадикалов уменьшается и снижается скорость обрыва.
Рис.3.2 ИВ
Величина наблюдаемого самоускорения колеблется в широких пределах и зависит от природы мономера, температуры, наличия инертного растворителя. В широких пределах изменяется и глубина полимеризации, при которой проявляется гель-эффект: метакрилат – 1 % (30 оС); метилметакрилат – 15 % (30 оС); стирол – 30 % (50 оС).
При гомогенном протекании радикальной полимеризации мономера в массе (в отсутствие растворителя) рассматривают четыре ее стадии:
Участок стационарной кинетики, когда скорость процесса и молекулярная масса полимера практически постоянные.
На второй стадии происходит существенное ускорение полимеризации и увеличение молекулярной массы полимера.
На третьей стадии молекулярная масса начинает уменьшаться, скорость уже не увеличивается, но еще остается высокой.
Наконец, процесс затухает на 4 стадии, что связано как с частичным исчерпанием мономера, так и с затвердеванием реакционной смеси.
Из-за гель-эффекта масштабное производство полимеров блочным способом (в массе) является нецелесообразным и опасным (может произойти неконтролируемый процесс резкого ускорения полимеризации с большим тепловыделением; проведение процесса в больших объемах может привести к выбросу или даже взрыву). В промышленности большинство полимеров получают в условиях, когда гель-эффект не проявляется – в растворе, эмульсии или суспензии.
Для получения прозрачных монолитных блоков ПММА или ПС больших размеров для полимеризации берут форполимер (5-10% р-р полимера в мономере, полученный на первой стадии синтеза, вовремя прервав ее), в систему вводят замедлители, а также специальные добавки, которые при повышенных температурах генерируют слабые ингибиторы (самый эффективный способ: чем больше выделяется тепла, тем больше образуется малоактивных радикалов, которые «гасят» часть макрорадикалов).
По мере дальнейшего течения реакции увеличивается скорость, которая у метилметакрилата достигает максимума при 60 % глубине полимеризации. После чего скорость реакции резко снижается. При этом образуется макрогель и скорость передвижения мономерных молекул в нем встречает большое сопротивление, и скорость полимеризации резко падает. В случае, когда реакция проводится при температуре, меньшей, чем температура стеклования, реакция практически прекращается, но она может быть доведена до конца введением инертного растворителя или повышением температуры.
Представленное объяснение в целом правильно (качественно) объясняет природу гель-эффекта. Для количественного объяснения изменения скорости полимеризации с увеличением глубины полимеризации учитывают различные факторы: изменение концентрации реагентов полимеризующейся среды, увеличение роли реакций передачи цепи, рекомбинации макрорадикалов и первичных радикалов, образование физической сетки перекрывающихся клубков макромолекул, образование надмолекулярных образований типа доменов и др.
С учетом таких особенностей протекания процесса полимеризации его можно разделить на четыре стадии:
При низких степенях конверсии скорость полимеризации описывается уравнением кинетики идеальной полимеризации. Mn и Mw – практически не изменяются.
При сохранении постоянной скорости инициирования происходит автоускорение полимеризации. Mn и Mw – увеличивается.
Автоускорение прекращается, скорость полимеризации остается высокой. Mn – увеличивается, Mw – уменьшается.
Реакция прекращается, не достигнув полного расходования мономера.
М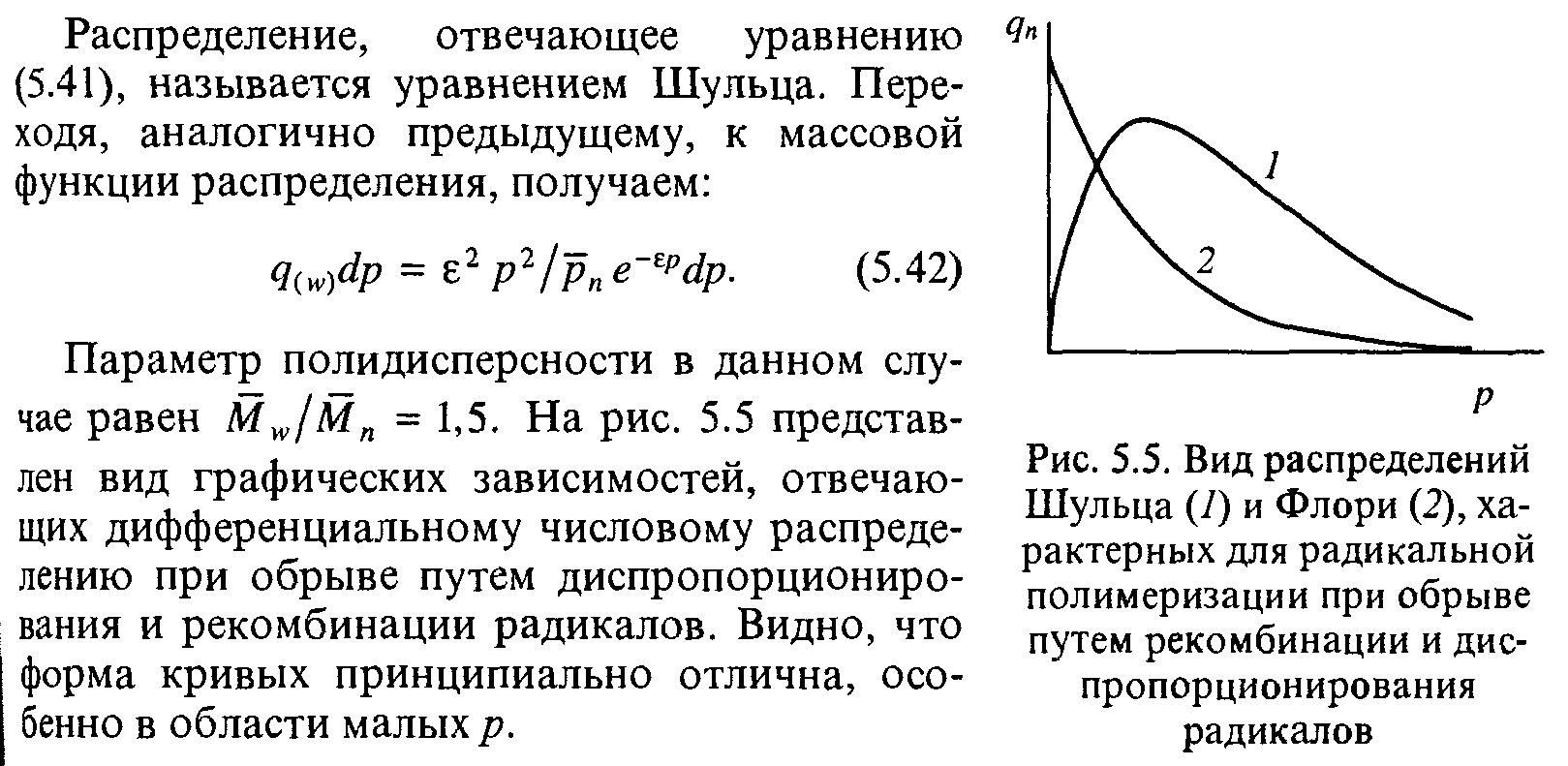 МР
полимера является суммой мгновенных
распределений фракций, образованных в
разные моменты времени. Это приводит к
его значительному уширению, которое
характеризуется коэффициентом
полидисперсности. Для ПЭВД этот
коэффициент в зависимости от условий
получения находится в интервале от 5 до
25, а для полистирола – от 2 до 6.
МР
полимера является суммой мгновенных
распределений фракций, образованных в
разные моменты времени. Это приводит к
его значительному уширению, которое
характеризуется коэффициентом
полидисперсности. Для ПЭВД этот
коэффициент в зависимости от условий
получения находится в интервале от 5 до
25, а для полистирола – от 2 до 6.
На рисунке представлен вид графических зависимостей, отвечающих дифференциальному числовому распределению при обрыве путем диспропорционирования и рекомбинации радикалов. Видно, что форма кривых принципиально отлична, особенно в области малых р.
В радикальной полимеризации широко используют полифункцион. инициаторы, мономеры, агенты передачи цепи, повторное участие к-рых в ходе полимеризации изменяет структуру полимера или кинетич. характеристики. Так, полиинициаторы способны придавать радикальной полимеризации кинетич. закономерности поликонденсации, из полифункцион. мономеров образуются сшитые полимеры, а введение полифункцион. агентов передачи цепи приводит к получению звездообразных полимеров.
|
|
|
(1) |
|
|
где kР – константа скорости роста цепи. Для исключения из этого уравнения концентрации промежуточного продукта (макрорадикала) исходят из того, что скорость изменения их концентрации равна разности скоростей инициирования WИ и скорости реакций обрыва цепей WО:
|
|
|
|
|
|
При стационарном режиме
|
|
|
|
|
|
откуда
|
|
|
|
|
|
Но скорость инициирования пропорциональна концентрации инициатора
|
|
|
|
|
|
Здесь
и далее kИ
является эффективной константой,
учитывающей, что при распаде инициатора
образуется два первичных радикала, а
инициирование идет с эффективностью
f:
![]()
Если скорость обрыва обусловлена только столкновением двух растущих макрорадикалов, то
|
|
|
|
|
|
Следовательно
|
|
|
|
|
|
и стационарная концентрация макрорадикалов будет равна
|
|
|
|
|
|
 .
.Подставляя найденное значение в уравнение (1) получаем
|
|
|
|
|
|

или
|
|
|
(2) |
|
|
где
|
|
|
|
|
|

Учитывая, что
|
|
, |
|
|
|
выражение (2) можно переписать следующим образом:
|
|
|
|
|
|
т.е. общая скорость полимеризации прямо пропорциональна концентрации мономера и квадратному корню из скорости инициирования (или квадратному корню из концентрации инициатора).
Средняя длина кинетической цепи зависит от соотношения скоростей реакции обрыва и роста цепи: чем больше вторая по сравнению с первой, тем больше молекул мономера присоединится к макрорадикалу к моменту обрыва цепи. Другими словами, длина кинетической цепи прямо пропорциональна скорости роста и обратно пропорциональна скорости обрыва цепи:
|
|
|
|
|
|
Для стационарного режима, когда WP = WO
|
|
|
|
|
|
При
отсутствии каких либо других элементарных
реакций, кроме инициирования роста и
обрыва, длина кинетической цепи
пропорциональна средней степени
полимеризации. При обрыве путем
рекомбинации
![]() ,
а при диспропорционировании –
,
а при диспропорционировании –
![]() .
Таким образом, в простейшем случае можно
отождествить длину кинетической цепи
со средней степенью полимеризации и
считать, что
.
Таким образом, в простейшем случае можно
отождествить длину кинетической цепи
со средней степенью полимеризации и
считать, что
|
|
|
|
|
|
Здесь и далее для упрощения выводов предполагается, что обрыв цепи идет только при помощи диспропорционирования.
Поскольку
![]() ,
то подставляя значение общей скорости
и скорости инициирования, получаем
,
то подставляя значение общей скорости
и скорости инициирования, получаем
|
|
|
|
|
|
 .
.или
|
|
|
(3) |
|
|
где
|
|
|
|
|
|
При наличии передачи цепи средняя степень полимеризации будет обратно пропорциональна сумме скорости обрыва цепи и всех скоростей передачи цепи (через мономер, растворитель, полимер, регулятор и т.д.):
|
|
|
|
|
|
Если проводить полимеризацию до небольшой глубины, то можно во многих случаях ограничится учетом передачи цепи только на мономер, тогда
|
|
|
|
|
|
Выражая концентрацию макрорадикала через скорость полимеризации и концентрацию мономера
|
|
|
|
|
|
получаем
|
|
|
|
|
|

или
|
|
|
(4) |
|
|
Если обрыв цепи происходит не только диспропорционированием, но и за счет рекомбинации, то
|
|
|
(4а) |
|
|
где – доля реакций обрыва цепи, проходящих путем диспропорционирования.
В
соответствии с уравнением (4),
экспериментально найденная зависимость
![]() от W[M]2
представляет собой прямую линию с
наклоном
от W[M]2
представляет собой прямую линию с
наклоном
![]() ,
отсекающую на оси
отрезок
,
отсекающую на оси
отрезок
![]() .
Если известно kР,
то легко можно вычислить kМ.
Прохождение прямой через начало координат
указывает на отсутствие передачи цепи
(kM = 0).
В этом случае уравнение (4) приобретает
вид:
.
Если известно kР,
то легко можно вычислить kМ.
Прохождение прямой через начало координат
указывает на отсутствие передачи цепи
(kM = 0).
В этом случае уравнение (4) приобретает
вид:
|
|
|
(4б) |
|
|

Наличие обратной пропорциональной зависимости между степенью полимеризации и общей скоростью становится особенно заметной при больших значениях отношения . В этом случае для получения полимера с большой степенью полимеризации процесс необходимо проводить с очень малой скоростью.
Рис.15Ш
Факторы, влияющие на скорость полимеризации.
Влияние температуры. Поскольку скорость полимеризации равна
|
|
, |
|
|
|
то температурная зависимость константы скорости полимеризации в соответствии с уравнением Аррениуса будет
|
|
|
|
|
|
 =
= ,
,где EР, EИ, EО – соответственно энергии активации реакций роста, инициирования и обрыва цепи; E – общая энергия активации; A, AР, AИ, AО – соответствующие предэкспоненциальные члены.
Откуда
|
|
|
|
|
|
Значения EР для большинства мономеров равно приблизительно 30 кДж/моль, EО – колеблется в пределах 12-20 кДж/моль, а EИ 120 кДж/моль (энергия активации распада бензоилпероксида). Поэтому повешение температуры значительно больше влияет на скорость инициирования, чем на роста и обрыва цепи. Естественно, с ростом температуры повышается общая скорость полимеризации.
Аналогично
для константы уравнения (3) для средней
степени полимеризации
![]()
|
|
=
|
|
|
|
Подставляя в уравнение в уравнение (3) и логарифмируя, получаем:
|
|
|
(5) |
|
|
Поскольку концентрация мономера и инициатора не зависят от температуры, дифференцируя уравнение (4) по температуре получим
|
|
|
|
|
|
Так,
как
![]() ,
то значение числителя меньше нуля.
Следовательно,
,
то значение числителя меньше нуля.
Следовательно,
![]() отрицательно и средняя степень
полимеризации падает с возрастанием
температуры. Такая зависимость
действительно наблюдается для ряда
систем. Например, для стирола,
метилметакрилата, винилацетата.
отрицательно и средняя степень
полимеризации падает с возрастанием
температуры. Такая зависимость
действительно наблюдается для ряда
систем. Например, для стирола,
метилметакрилата, винилацетата.
Рис.14Ш
При
фотополимеризации, когда EИ
близко к нулю и
![]() ,
тогда
положительно и средняя степень
полимеризации растет с температурой
(в предположении, что передача цепи
отсутствует). По этой же причине скорость
фотополимеризации слабо зависит от
температуры.
,
тогда
положительно и средняя степень
полимеризации растет с температурой
(в предположении, что передача цепи
отсутствует). По этой же причине скорость
фотополимеризации слабо зависит от
температуры.
Анализ уравнения зависимости средней степени полимеризации от константы передачи цепи на растворитель
показывает, что энергия активации передачи цепи на растворитель составляет 50-90 кДж/моль и значительно выше энергии активации реакции роста цепи. Поэтому при повышении температуры скорость передачи цепи растет быстрее, чем скорость роста цепи. Следовательно, при наличии передачи цепи молекулярный вес полимеров снижается.
СКОРОСТЬ ПОЛИМЕРИЗАЦИИ И ФАКТОРЫ, КОТОРЫЕ НА НЕЕ ВЛИЯЮТ
Общая скорость радикальной полимеризации V равна скорости исчезновения мономера при его взаимодействии с растущим радикалом. Число мономерных молекул, участвующих в инициировании, очень мало, по сравнению с количеством молекул, участвующих в реакциях роста цепи, поэтому можно считать, что общая скорость приблизительно равна скорости роста цепи Vр:
![]()
Непосредственно концентрацию радикалов измерить сложно, т. к. она очень мала. Поэтому необходимо выразить скорость процесса через другие величины. Для исключения из этого уравнения концентрации макрорадикалов исходят из того, что скорость изменения их концентрации равна разности скоростей инициирования VИ и скорости реакций обрыва цепей VО. На начальных стадиях превращения радикальная полимеризация, как правило, протекает с постоянной скоростью, что связано с выполнением условия квазистационарности (постоянства концентрации активных центров) – все время одни радикалы возникают, другие исчезают, рекомбинируя, и их концентрации становятся стационарными, т.е. неизменными во времени (принцип Боденштейна–Нернста). В итоге скорость инициирования равна скорости обрыва: vи = vо.
В общем виде скорость радикальной полимеризации описывается уравнением:
 ,
,![]()
![]()
![]()
где
Т.е. общая скорость полимеризации прямо пропорциональна концентрации мономера и квадратному корню из скорости инициирования (или квадратному корню из концентрации инициатора).
Типичная кинетическая кривая, описывающая конверсию мономера (т. е. превращение мономера в полимер в результате полимеризации) в зависимости от времени, имеет S-образный вид (рис. 1.2). Как видно из рис. 1.2, на кривой можно выделить пять участков по значениям скоростей основной реакции превращения мoномера в полимер в результате полимеризации: 1) участок ингибирования, где концентрация свободных радикалов мала и они не могyт начать цепной процесс полимеризации; 2) участок ускopeния полимеризации, гдe начинается основная реакция превращения мономера в полимер, причем скорость растет; 3) участок cтaциoнарного состояния, где происходит полимеризация основнoго кoличества мономера при постоянной скорости (прямолинейная зависимость конверсии от времени); 4) участок замедления полимеризации, где скорость реакции уменьшается в связи с убылью содержания свободногo мономера; 5) прекращение основной peaкции после исчерпания вceгo количества мономера.

Влияние количества инициатора
Зависимость скорости полимеризации и средней молекулярной массы от концентрации инициатора была рассмотрена выше. Скорость изменяется прямо пропорционально, а средняя молекулярная масса – обратно пропорционально корню квадратному из концентрации инициатора.
При отсутствии реакции передачи цепи средняя степень полимеризации пропорциональна скорости реакции роста цепи и обратно пропорциональна скорости реакции обрыва цепи:При наличии передачи цепи средняя степень полимеризации будет обратно пропорциональна сумме скорости обрыва цепи и всех скоростей передачи цепи (через мономер, растворитель, полимер, регулятор и т.д.):
![]()
Если проводить полимеризацию до небольшой глубины, то можно во многих случаях ограничиться учетом передачи цепи только на мономер, тогда
![]()
Выражая концентрацию макрорадикала через скорость полимеризации и концентрацию мономера
![]() ,
,
получаем

или
![]()
Если обрыв цепи происходит не только диспропорционированием, но и за счет рекомбинации, то в первое слагаемое вводится множитель 1/2:
![]() ,
,
где – доля реакций обрыва цепи, проходящих путем диспропорционирования.
![]()
При обрыве рекомбинацией перед скоростью обрыва вводится множитель ½.
Т.е. степень полимеризации и, следовательно, средняя молекулярная масса полимера при свободнорадикальной полимеризации обратно пропорциональны квадратному корню из концентрации инициатора.
С ростом концентрации инициатора растет и число радикалов, образующихся в системе. Эти радикалы реагируют с большим числом молекул мономера и тем самым увеличивают скорость их превращения в растущие макрорадикалы. Однако различные инициаторы, при одном и том же мономере действуют те одинаково. По-разному также ведет себя один и тот же инициатор по отношению к различным мономерам. Например, скорости полимеризации бутадиена, стирола, и акрилонитрила в присутствии 1 % бензоилпероксида относятся друг к другу как 1 : 500 : 100 000, а в присутствии 1 % диазоаминобензола – как 1 : 3 : 25.
Такие различия некоторые авторы объясняют комплексообразованием инициаторов и мономеров, с последующим образованием первичного радикала. Однако правильнее и лучше такое явление объясняется дезактивацией первичных радикалов. Этот вопрос был рассмотрен ранее.Однако при общем увеличении концентрации радикалов растет и вероятность их столкновения друг с другом, т.е. обрыва цепи полимеризации, что приводит к снижению средней молекулярной массы полимера.
Влияние концентрации мономера
В соответствии с формулами (2) и (3) Уменьшение концентрации мономера ведет к снижению скорости полимеризации и средней степени полимеризации. Одновременно усиливается роль передачи цепи на растворитель, что вызывает дополнительное снижение средней молекулярной массы. При этом существенную роль играет природа растворителя, поскольку скорость передачи цепи на растворитель не одинакова для различных растворителей.
Температура
Суммарная Еа процесса полимеризации связана с энергиями активации элементарных стадий: Еа = Ер + ½ Ерасп – ½ Ео.
Поскольку Ерасп обычно употребляемых инициаторов заключены в пределах 125-150 кДж/моль, а величины Ер и Ео, соответственно, в пределах 20-40 кДж/моль и 2-20 кДж/моль, суммарная энергия активации полимеризации наиболее распространенных мономеров составляет Е ~ 80-90 кДж/моль. Суммарная энергия активации полимеризации связана с энергиями активации элементарных реакций соотношением:
Используя известные значения энергетических параметров полимеризации, можно сделать следующие выводы о влиянии температуры на скорость и степень полимеризации:
1. Поскольку Еи обычно употребляемых инициаторов заключены в пределах 125-150 кДж/моль, а величины Ер и Ео, соответственно, в пределах 20-40 кДж/моль и 2-20 кДж/моль, суммарная энергия активации полимеризации наиболее распространенных мономеров составляет Е ~ 80–90 кДж/моль. Это означает, что скорость полимеризации возрастает в 2-3 раза при повышении температуры на 10 оС, что примерно соответствует правилу Вант-Гоффа. Применение окислительно-восстановительных систем снижает Е примерно вдвое. Величина суммарной энергии активации степени полимеризации отрицательна и обычно близка к -60 кДж/моль, поэтому молекулярная масса полимера весьма заметно понижается с ростом температуры.
2. При фотохимической полимеризации Еи = 0, поэтому скорость и степень полимеризации возрастают с ростом температуры вследствие положительной величины суммарной энергии активации, близкой к 20 кДж/моль.
3. Энергия активации реакции передачи цепи заметно превосходит энергию активации реакции роста, обычно Ер - Es = -(20 - 60) кДж/моль. Это означает, что при полимеризации в их присутствии повышение температуры приводит к уменьшению степени полимеризации.
Во всех случаях, за исключением фотополимеризации, повышение температуры полимеризации снижает молекулярный вес полимера; усиливается роль побочных процессов, требующих сравнительно высоких энергий активации и слабо выраженных при низких температурах: аномальные присоединения "голова к голове", полимераналогичные превращения, деструкционные процессы, что нарушает регулярность структуры и ухудшает свойства полимеров.
При более низких температурах ММР сужается, так как подвижность макрорадикалов уменьшается и снижается доля их столкновений при малых степенях полимеризации, что сокращает долю низкомолекулярной фракции.
Молекулярная масса растет вследствие уменьшения вероятности обрыва реакционной цепи при уменьшении подвижности макрорадикалов.
Снижение температуры положительно сказывается на регулярности чередования звеньев в цепи.
Снижается или полностью отсутствует разветвленность макромолекул.
Однако при более низких температурах снижается общая скорость полимеризации, и для поддержания ее на уровне оптимальной производительности оборудования приходится применять окислительно-восстановительные системы для инициирования распада молекул инициатора или специально вводить активирующие этот распад добавки (т.н. промоторы полимеризации).
Обычно скорость полимеризации возрастает в 2–3 раза при повышении температуры на 10 оС (что примерно соответствует правилу Вант-Гоффа). Рост температуры увеличивает скорость инициирования полимеризации, так как облегчает распад на радикалы инициаторов и их реакцию с молекулами мономера. Вследствие большей подвижности малых радикалов с повышением температуры увеличивается вероятность их столкновения друг с другом (обрыв цепи путем диспропорционирования или рекомбинации) или с низкомолекулярными примесями. В результате молекулярная масса полимера в целом уменьшается (средняя степень полимеризации понижается с ростом температуры), а также увеличивается доля низкомолекулярных фракций в полимере (расширяется ММР). Возрастает число побочных реакций, приводящих к образованию разветвленных молекул, увеличивается нерегулярность при построении цепи полимера вследствие возрастания доли типов соединения мономера «голова к голове» и «хвост к хвосту».
Давление
В отличие от других способов ускорения полимеризации, При повышении давления до 100 МПа и выше одновременно увеличиваются и скорость полимеризации, и средняя молекулярная масса. При температуре 333 К с ростом давления от 0,1 до 600 МПа наблюдается логарифмическая зависимость скорости полимеризации стирола, инициированная пероксидом бензоила и при 300 МПа она в 7 раз выше, чем при нормальном давлении. Одновременно молекулярная масса увеличивается в 3 раза. При дальнейшем увеличении давления она возрастает незначительно, приближаясь к некоторому пределу.Возрастание скорости объясняют, тем, что сильное сжатие сближает реагирующие частицы и уменьшает длину свободного пробега молекул мономера. Кроме того, с ростом давления увеличивается вязкость системы и уменьшается скорость рекомбинации макрорадикалов.
В пользу такого объяснения свидетельствует увеличение с ростом давления константы роста цепи и снижение константы реакций обрыва цепи.
Рис.16Ш
Следует также отметить, что некоторые мономеры, не полимеризующиеся в обычных условиях, при повышенных температурах полимеризуются со значительным выходом полимеров. При высоких давлениях повышается предельная температура полимеризации, и тем самым осуществляется полимеризация мономеров с небольшой теплотой полимеризации, что в обычных условиях термодинамически не возможно. (см. стр.132Ш)