

8.5.3. Теорія перехідних станів
Хоч ідея про перехідний стан була запропонована ще Кекуле (див. розділ 7.3.4.2) в 1858 р., теорія перехідних станів стала розроблятися десь із середини 30-х років, але вона виникла не на пустому місці.
Сванте Ареніус в 1889 р. розробив теорію залежності швидкості реакції від температури, увів поняття “активних молекул” і енергії активації Еа та вивів рівняння залежності константи k швидкості реакції від фактора А частоти зіткнення реагуючих молекул і енергії активації (у логарифмічному вигляді):
lnk = lnA – Ea/RT.
Еа – енергія, яку повинні мати реагуючі молекули при зіткненні, щоб подолати потенціальний бар’єр реакції (див. рис. 8.1).
Потенціальна енергія
Енергія активації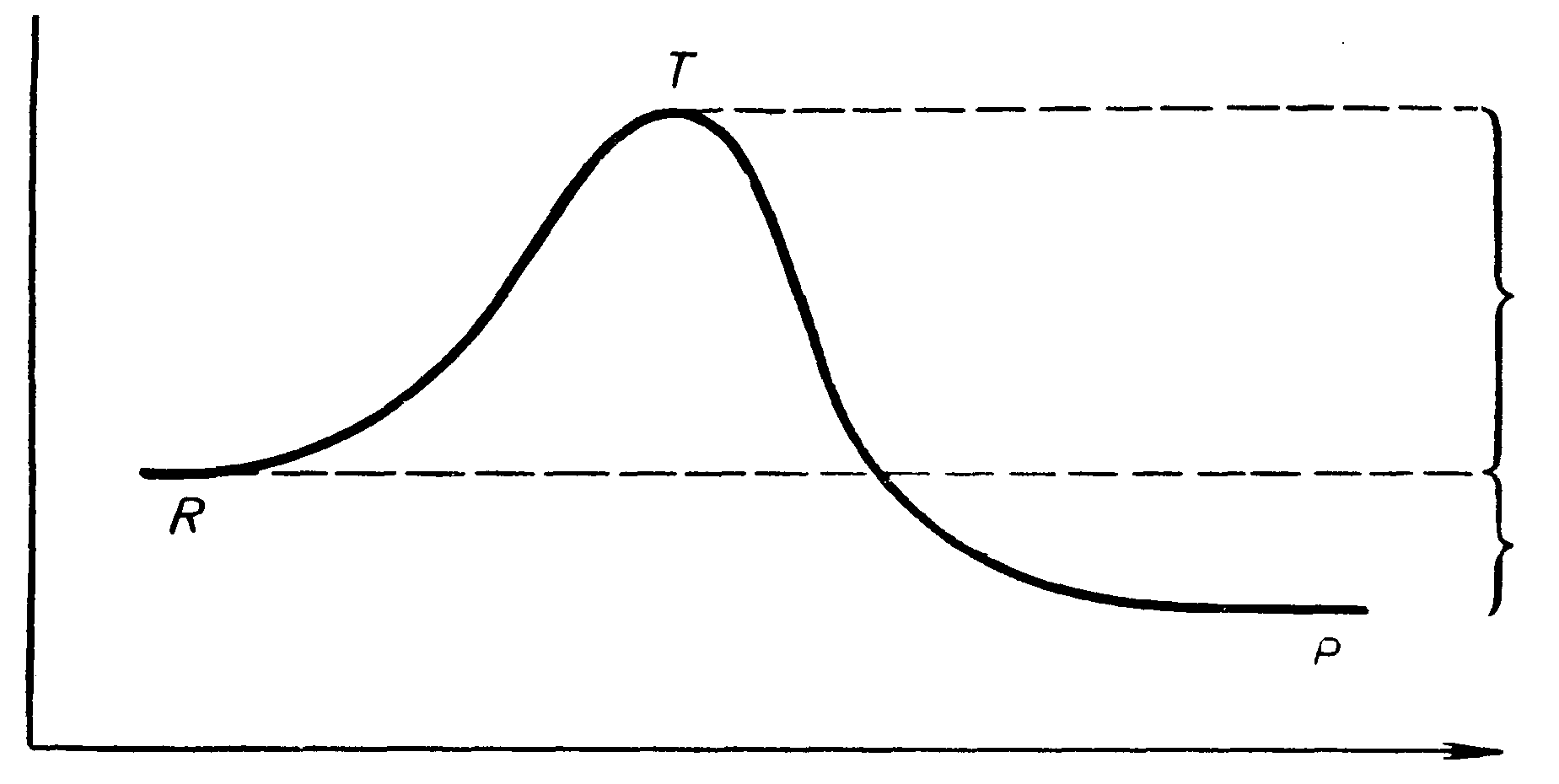
Шлях реакції
Енергія реакції
Необхідно відзначити, що структуру перехідних станів не можна розглядати відокремлено від механізму реакції, як це робить Г.В.Биков, автор “Історії органічної хімії”, оскільки перехідний стан є невід’ємним елементом механізму, бо від того, яким шляхом утворюється перехідний стан, залежить природа та будова продуктів реакції. Величезний неоцінимий внесок у розв’язання проблеми встановлення механізмів органічних реакцій кінетичними методами зробили два згадані вище англійські фізико-хіміки Хіншельвуд та Інгольд (розділи 8.4.2 та 8.5.1).
Ще в 30-ті роки ХХ ст. були зроблені спроби дослідження просторової будови (стереохімії) перехідних станів. Так, Інгольд та Хьюз, вивчаючи кінетику та стереохімію бімолекулярних реакцій нуклеофільного заміщення при насиченому атомі вуглецю, дійшли висновку, що в цьому випадку перехідний комплекс, позначений індексом , утворюється за рахунок тилової атаки нуклеофіла Nu:

 R2
R2
R1
N u + R1R2R3CX Nu C X NuCR1R2R3 + X
R
В цьому комплексі три замісники, R1 , R2 та R3, лежать в одній площині, а атакуючий нуклеофіл та група Х, що відщеплюється, знаходяться по різні сторони від цієї площини. Унаслідок такої атаки відбувається обернення конфігурації асиметричної структури. Таке обернення конфігурації тепер називають Вальденівським по імені хіміка Пауля Вальдена (1863 – 1957), який відкрив це явище в 1898 році. В наступні десятиліття багатьма дослідниками було показано, що в залежності від умов реакції та будови реагентів атака нуклеофіла на насичений атом вуглецю може також призвести до збереження конфігурації (фронтальна атака зі сторони знаходження групи, що відщеплюється) та до рацемізації внаслідок фронтальної та тилової атак у рівній мірі. Цілком зрозуміло, що тут просто змінювався механізм реакції та напрямок атаки.
В 1960 - 1966 роки Олег Олександрович Реутов (нар. в 1920 р.) разом з Іриною Петрівною Білецькою (нар. в 1933 р.), вивчаючи реакції електрофільного заміщення за участю ртутьорганічних сполук, показали, що конфігурація асиметричного атома в перехідному стані зберігається, а сам перехідний стан має вигляд чотирьохчленного циклу:

 Hg
Hg
C X2
Hg
Причім, тут, як і при нуклеофільному заміщенні, в залежності від механізму реакції має місце як збереження, так і обернення конфігурації. Звертає на себе увагу припущення про утворення циклічної будови перехідного стану.
Академік, перший ректор Донецького університету і перший завідувач кафедри органічної хімії в цьому університеті Леонід Михайлович Литвиненко (1921 – 1983) із співробітниками показали, що утворення циклічних перехідних станів є необхідною умовою проявлення біфункціонального каталізу в реакціях нуклеофільного заміщення при ненасиченому атомі вуглецю в неполярних середовищах. Подальші дослідження багатьох учених кінетики реакцій у неполярних середовищах наводять на думку, що утворення циклічних перехідних станів у таких умовах запобігає суттєвому розділенню зарядів по ходу реакції, завдяки чому сама реакція і відбувається з помітною швидкістю.
В 1955 році американський фізико-хімік Джордж Сімс Хемонд (нар. в 1921 р.) висунув постулат, виходячи з якого, можна вирішувати, вихідні реагенти чи кінцеві продукті реакції є хорошою моделлю перехідних станів: для високо екзотермічних реакцій вони будуть реагентоподібними, для високо ендотермічних реакцій перехідні стани будуть продуктоподібними.
Квантовохімічні розрахунки можна застосовувати для оцінки вірогідності існування тих чи інших проміжних продуктів реакції, в тому числі і перехідних станів. До таких проміжних продуктів належать, наприклад, карбокатіони, які часто мають “некласичну” будову. Так, проведені в 1964 році ретельні розрахунки Гофмана (Hoffmann R.) методом молекулярних орбіталей дозволили зробити висновок, що для катіона С2Н5+, що утворюється при приєднанні Н+ до етилену, має будову:
Н

 +
+
С С ,
де два електрони “розмазані” по трьох зв’язках.
Утворення катіона С6Н7+ шляхом приєднання протона до молекули бензолу виявилося найбільш вигідним у разі наближення Н+ вподовж одного з існуючих зв’язків СН та найменш вигідним при наближенні вподовж осі симетрії, яка перпендикулярна площині кільця. Такі розрахунки відкрили можливість як для оцінки енергії активації, так і для висновків про кращу геометричну будову перехідних станів.
В 1983 році Менджер* висунув гіпотезу, що реагент може рухатися не по одній чітко визначеній траєкторії, а по багатьом, які розміщені всередині реакційної вирви (вікна):





 В
АХ
В
АХ
Ця гіпотеза перехідного стану, який можна назвати багато направленим, дозволяє пояснити деякі невідповідності теоретичних уявлень експериментальним фактам. Так, реакційна постійна у рівнянні Гаммета [ lg (kR/kH) = R ], яка по суті є характеристикою положення
* Menger F.M. Directionality in organic reaction in solution//Tetrahedron. – 1983. – Vol. 39, № 7. – р. 1013 – 1040.
перехідного стану на шляху від реагентів до продуктів і тому принципово не повинна бути постійною величиною, насправді зберігає постійне значення. Ця величина є середньостатистичною макроскопічною, то її усереднене значення мало змінюється при дуже широкій зміні параметрів реакційного вікна та розподілу траєкторій за енергією всередині вікна. Інша невідповідність – малі значення кінетичних ізотопних ефектів у порівнянні з передбачуваними теорією його значеннями, теж пояснюється макроскопічною природою цих ефектів, які залежать від тих же вказаних вище причин.
В 1965 році Вудворд (Woodward R.B.) і Хофман запропонували правило збереження симетрії реагуючих орбіталей для узгоджених одно стадійних реакцій типу Дільса-Альдера, яке випливає з квантовохімічних методів: збурення молекулярних орбіталей та граничних орбіталей. Це правило, як і квановохімічні методи, дозволяють пояснити не лише можливість таких реакцій, а й їх високу стерео специфічність.
Луїс Плек Гаммет (1894 – 1987) – американський фізико-хімік, професор Колумбійського університету. Роберт Бернс Вудворд (1917 – 1979) - американський хімік-органік, професор Гарвардського університету, Нобелівська премія за 1965 р. Роалд Хофман (р.н. 1937) – американський хімік, професор Корнеллського університету, Нобелівська премія за 1981 р. |
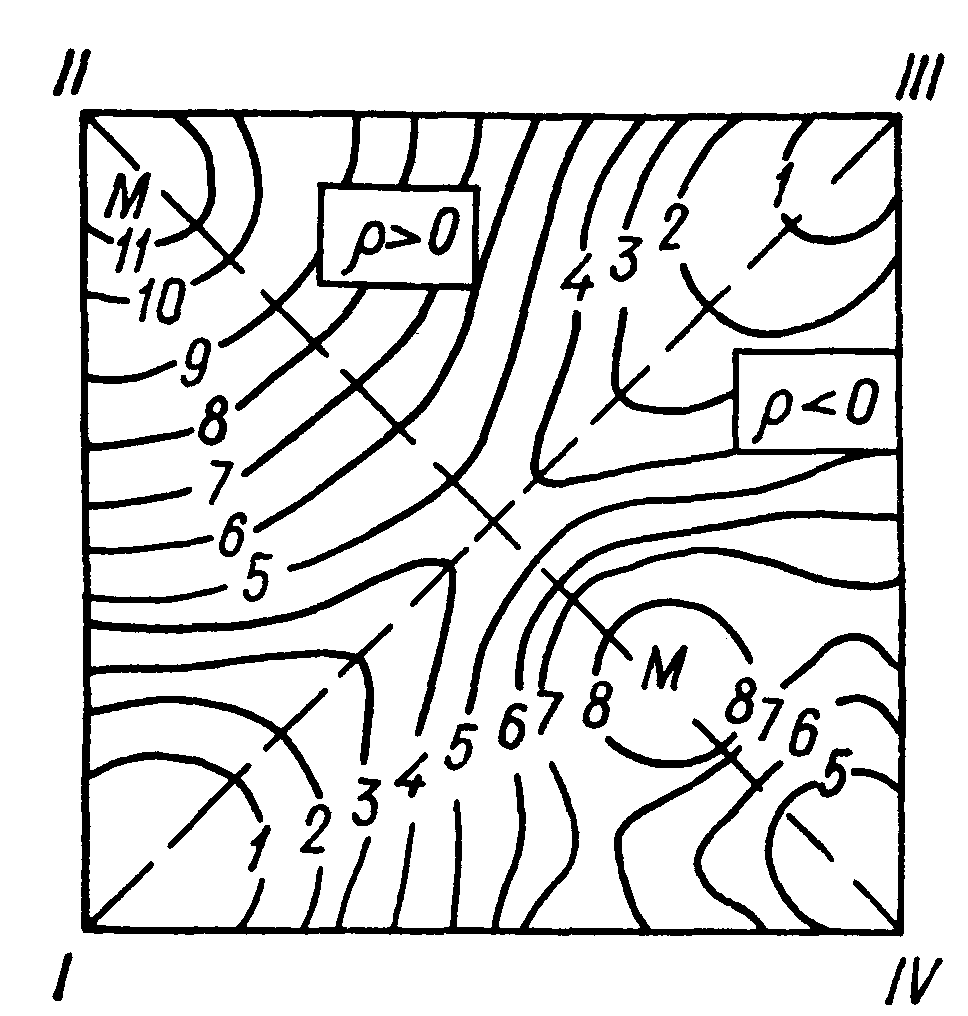
Уявлення Хемонда про структуру перехідних станів були розвинені Торнтоном (Tornton T.R.) та О’Ферралом (O’Ferrall R.A.M.) в 1967 – 1970 роках. Пізніше до них приєднався професор Петербурзького
університету Дніпровський О.С. Для опису шляхів реакцій вони запропонували трьохмірні діаграми, приклад такої діаграми показаний на рис. 8.2 для реакцій заміщення біля насиченого атома вуглецю. Дві
взаємоперпендикулярні координати, що лежать в площині паперу, відповідають змінам довжин двох зв’язків у реагуючій системі, а третя координата відповідає вільній (потенційній) енергії цієї системи. По суті справи маємо схематичне зображення поверхні потенційної енергії, подібне географічній карті, на якому суцільні лінії відображають рівні однакової енергії (на географічній карті такі лінії відповідають однаковій висоті земної поверхні над рівнем моря). Такі діаграми показують не лише всі можливі механізми реакції, яка розглядається, а й передбачає напрямки змін положення перехідного стану на цій діаграмі в залежності від змін потенційної енергії реагентів, продуктів реакції та проміжних продуктів реакції, координати яких розміщено у вершинах квадрата діаграми.
Таким чином, треба підкреслити, що найбільший успіх у наукових досягненнях із початку ХХ століття припадає на ту наукову галузь,
rC-Nu
Nu
+ R3C+
+ X
rC-X
Nu
+
R3CX
т
Nu(CR3)X
NuCR3
+ X