

Завдання 3. Визначення розмірно-масових характеристик хімічного складу і показників якості риби фізичним і фізико-хімічним методами
Визначення розміру та маси риби. За розміром (довжиною) та масою більшість видів риб підрозділяють на крупну та дрібну (ГОСТ 1368).
Харчова цінність крупних особин одного і того ж сімейства (виду) вище, ніж дрібних.
У промисловості та торгівлі розмір риби визначають у відповідності із правилами рибальства та нормативними документами.
Промислову довжину риби змірюють по прямій лінії від початку (вершини) рила до початку середніх променів хвостового плавця (мал.11).
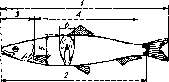
1 – загальна (зоологічна) довжина; 2 -
довжина тушки (промислова довжина);
3 - довжина голови; 4 - довжина тушки;
5 - висота тіла; 6 - товщина тіла
При визначенні довжини (зоологічної, промислової та ін.) рибу слід укласти на рівну поверхню (стіл, дошку). Довжину вимірюють за допомогою металевої лінійки з міліметровими діленнями.
Довжину неподіленої та патраної із головою риби вимірюють по прямій лінії - від вершини рила до основи середніх променів хвостового плавника.
Довжину обезголовленої та патраної обезголовленої риби вимірюють по прямій лінії від краю головного зрізу на рівні хребта до основи середніх променів хвостового плавця.
Довжину спинки (балика) вимірюють по прямій лінії від краю головного зрізу на рівні хребта до основи середніх променів хвостового плавця.
Риби, не перераховані у стандарті за найменуваннями, за довжиною та масою не підрозділяють. Окремі види риб (густера, голавль та ін.) відносять до дрібниці (першої, другої та третьої груп) та за довжиною та масою не поділяють.
Масу риби необхідно визначати поштучним зважуванням усіх екземплярів, що входять у відібрану пробу.
Визначення масової частки води. У гідробіонтах та продуктах, вироблених з них, форми зв'язку води з іншими компонентами (наприклад, з білками) різні. Розрізняються також стани та хімічні склади досліджуваних видів сировини та продукції. Усе це зумовило розробку різних методів визначення води, у тому числі: висушуванням зразка при 100-105 та 130 °С; відгоном з парами розчинників жиру (метод Дина та Старка); висушуванням інфрачервоними променями та ін.(ГОСТ 7636).
У виробничих лабораторіях найчастіше масову частку води в різних гідробіонтах та продуктах, вироблених з них, визначають висушуванням наважки зразку при температурі 100-105 °С. Сутність методу, підготовка до аналізу та техніка його проведення описані у ГОСТ 7636(п. 3.3.1) та в спеціальній літературі.
Визначення масової частки жиру. Більшість фізико-хімічних методів, які вживають для визначення кількості жиру, відносяться до так званих двоступінчатих, що передбачають витягання жиру з об'єкту (I ступінь) та кількісне визначення його (II ступінь).
Для витягання жиру використовують різні органічні розчинники - бензин, петролійний ефір, сірчаний ефір, ацетон, хлороформ, монобром, монохлорнафталін, трикрезилфосфат та ін.
Слід мати на увазі, що гідрофобні розчинники (петролійний ефір, бензин та ін.) витягають разом із гліцеридами декілька менше супутніх їм речовин, причому виділення їх відбувається селективно. Швидше виділяються гліцериди та повільніше фосфатиды, вільні жирні кислоти та продукти окислення. У зв'язку з цим при застосуванні гідрофобного розчину процес витягання жиру проходить повільно (упродовж 2-3 діб).
Для прискорення та більш повного виділення гліцеридів та супутніх їм речовин із аналізуємого об’єкту рекомендується використовівати гідрофільні розчинники (метиловий та етиловий ефіри та ін.) або суміш гідрофобних та гідрофільних розчинників(бінарні розчинники).
До найбільш часто вживаних методів визначення ліпідів відносяться екстракційно-вагові (метод Сокслета, метод визначення ліпідів по знежиреному залишку, метод ВНИИ-КОПа - метод Блая та Дайера, модифікований Інститутом харчування Російської Академії медичних наук) та екстракційно-рефрактометричний методи.
Методи, підготовка до аналізу та техніка їх виконання описані в ГОСТ 7636(п. 3.7) та у відповідній літературі.
Визначення масової частки азоту. Методи визначення азоту у сировині, напівфабрикатах та готовій продукції підрозділяють на дві групи: що передбачають мінералізацію (спалювання) наважки досліджуваного зразку; не передбачаючі спалювання наважки.
У лабораторіях рибопереробних підприємств, учбових та науково-дослідних галузевих інститутів найчастіше використовують методи, що передбачають мінералізацію наважки зразка.
Методи обгрунтовані на окисленні органічної речовини досліджуваного зразка при спалюванні його у сірчаній кислоті у присутності каталізатора, відгоні аміаку, що утворюється, парою, уловлювання його розчином сірчаної кислоти та визначенні вмісту азоту методом титрування.
Загальний азот Nоб визначають у вигляді аміаку NH3.
Утворившийся при розкладанні (окисненні) органічної речовини проби концентрованої гарячою сірчаною кислотою H2SO4 сульфат амонія (NH4) 2SO4 руйнується концентрованим лугом.
Отриманий аміак відганяють парою у титрований 0,1 н. розчин сірчаної кислоти та надлишок кислоти відтитровують 0,1 н. розчином лугу.
Реакції протікають за рівняннями
RCHNH2COOH + H2SO4 -> СО2 + SO2 + Н2О + NH3;
Аміак
2NH3 + H2SO4 -> (NH4)2SO4;
Сульфат амонію
(NH4)2SO4 + 2NaOH -> 2NH3 + 2Н2О + Na2SO4.
Аміак, що утворився, поглинається титрованим розчином сірчаної кислоти:
2NH3 + H2SO4 -> (NH4)2SO4 + H2SO4.
Надлишок
Надлишок сірчаної кислоти відтитровують гідроксидом натрію та за кількісті пов'язаної кислоти обчислюють кількість поглинаючого аміаку або кількість азоту, що відповідає йому.
Загальний азот визначають макрометодом, макрометодом із селеновою сумішшю та напівмікрометодом.
Методи, підготовка до аналізу та техніка їх проведення описані в ГОСТ 7636 (пп. 8.9.1 - 8 9.4) та в спеціальній літературі.
Деякі з методів досить короткочасні. Прискорення аналізу досягається шляхом раціонального підбору кількісного та видового складу основних реагентів та каталізаторів1, а також поєднання окремих операцій (наприклад, мінералізації, відгону та уловлювання аміаку) та зміни техніки їх проведення (наприклад, заміна титрування спектрофотометричним аналізом).
1 Для прискорення процесу спалювання рекомендується додавати : каталізаторну суміш, що складається з 1,9массової частини пероксидисульфата (персульфату) калію K2S2O8, 0,05 частини CuSO4 та 0,05 частини селену Se, пероксид водню; червоний оксид ртуті (~ 0 2 г) разом із сульфідом калію (~0,5 г) та інші каталізатори.
Кількість білку Б (у %) розраховують за рівнянням
Б = Nобщ · 6,25,
де Nобщ - вміст загального азоту, %; 6,25 - коефіцієнт перерахунку азоту у білок (100/16 = 6,25); значення коефіцієнту для різних видів гідробіонтів та продуктів, що виробляються з них, різноманітно та залежить від вмісту білку.
Визначення білкового азоту. Метод заснований на здатності білкових речовин утворювати із гідроксидом міді Си(ОН)2 з'єднання, не розчинні навіть у гарячій воді. Кількість азоту в отриманому осаді визначають макрометодом або іншим способом.
Для визначення вмісту азоту істинних білків (білковий азот) відважують 0,5-1,0 г (з похибкою не більше 0,01 г) тонко подрібненого в ступці досліджуваного матеріалу, поміщають його в термостійку хімічну склянку місткістю 100-150 см3, додають 50 см3 дистильованої води та нагрівають до кипіння. До нагрітої маси підливають 25 см3 розчину мідного купоросу (сульфату міді) (60 г CиSO4 · 5Н2О розчиняють у 1000 см3 дистильованої води) та при постійному перемішуванні підливають 25 см3 розчину NaOH (12,5 г NaOH розчиняють у 1000 см3 дистильованої води).
Після відстоювання суміші рідину обережно зливають декантацією через паперовий фільтр, а осад в склянці промивають кілька разів гарячою дистильованою водою, зливаючи промивні води через той же фільтр. Промивання проводять до тих пір, поки фільтрат не перестане давати реакцію на H2SO4(проба із хлоридом барію).
Промитий осад кількісно переносять на фільтр, висушують і разом із фільтром спалюють у колбі К’ельдаля1.
1 При визначенні білкового азоту в м'ясі жирних риб зібраний на фільтрі осад після висушування слід промити петролійним ефіром та знову під-сушують. Видалення жиру полегшує спалювання осаду із фільтром.
Усі подальші операції, починаючи із спалювання проби, виконують так само, як і при визначенні загального азоту.
Паралельно проводять холостий дослід в тих же умовах, але без наважки, що дозволяє встановити вміст азоту у фільтрі та реактивах. Результати контрольного досліду враховують у розрахунку.
Зміст істинних білків у досліджуваному матеріалі визначають шляхом множення отриманої кількості азоту на коефіцієнт 6,25.
Визначення вмісту небілкового азоту (азот пептонів, амінокислот). Небілкові азотвмісні речовини не осідають під дією осаджувачів білків (наприклад, трихлороцтовою кислотою - ТХК). На цій властивості заснований метод їх визначення.
Наважку фаршу 50 г, відважене із похибкою не більше 0,01 г, розтирають у ступці з 100-150 см3 дистильованої води, переносять в мірний циліндр із пришліфованою пробкою та доводять об'єм суміші до 250 см3. Суміш збовтують в апараті впродовж 1 год із швидкістю 50-60 коливань у хвилину. Отриману масу профільтровують спочатку через один шар марлі, а потім
через чотири шари. Від фільтрату відбирають піпеткою 100 см3, поміщають в колбу місткістю 250-300 см3 та підливають невеликими порціями при збовтуванні вмісту 25 см3 20%-ого розчину трихлороцтової кислоти (ТХК). Після 30-хвилинного відстоювання рідину фільтрують через сухий складчастий фільтр. У 5-10 см3 фільтрату(залежно від очікуваної кількості небілкового азоту у досліджуваному продукті) визначають вміст азоту одним із вказаних вище методів.
При підрахунку результатів необхідно враховувати розведення наважки у ході аналізу.
Вміст небілкового азоту виражають у відсотках або міліграмах на 100 г продукту (мг/100 г).
Фільтрат, що залишився від визначення небілкового азоту описаним способом, може бути використаний для визначення азоту амінокислот методом формольного титрування(за Зеренсоном) або газометричним методом (за Ван-Слайком).
Визначення азоту амінокислот методом формольного титрування (за Зеренсоном). Амінокислоти та поліпептиди - амфотерні електроліти, тобто речовини, що несуть протилежні за знаком електричні заряди. У водному розчині вони одночасно проявляють властивості кислот та лугів. Їх вільні аміногрупи (NH2), що мають лужні властивості, нейтралізуються у водному середовищі вільними карбоксильними групами (СООН), що мають кислий характер.
При введенні формаліну у водні розчини амінокислот амінова група останніх зв'язується з ним, внаслідок чого її основні властивості зникають.
NH2 H N= CH2
RCH + С = О RCH + Н2О
СООН H СООН
Амінокислота Формалін Метиленаміно-
кислота
Протікає реакція з утворенням похідних типу метиленамінокислоти, яка є сильною кислотою,



Це дозволяє відтитровувати лугом вільні карбоксильні групи, що залишилися, та непрямим шляхом визначати кількість амінних груп.
N= CH2 N= CH2
RCH + КОН RCH + Н2О
СООН СООК
При титруванні лугом цього похідного реакція протікає за рівнянням
Проведення аналізу. 50 г досліджуваної речовини (фаршу) розтирають у ступці з 100-150 см3 дистильованої води, переносять в мірний циліндр з пришліфованою пробкою та доводять об'єм суміші до 250 см3. Суміш збовтують на апараті упродовж 1 год із швидкістю 50-60 гойдань у хвилину. Отриману масу (суспензію) відтитровують спочатку через один шар марлі, а потім через чотири шари та осаджують білки ТХК (25 см3 20%-ого розчину CCI3CO2H на 100 см3 фільтрату). Відфільтрувавши осад, відбирають 100 см3 фільтрату та додають до нього для осадження фосфатів та карбонатів 2 г сухого BaCl2 і 2 н. розчин NaOH до сильно лужної реакції на лакмус.
Після 15-хвилинного відстоювання рідину фільтрують через паперовий складчастий фільтр в колбу із притертою пробкою (для оберігання розчину від взаємодії з вуглекислим газом повітря). Відбирають в три колби з притертой пробкою по 25 см3 фільтрату. У одну з колб (№ 1) вносять 10 крапель розолової кислоти та проводять нейтралізацію розчину спочатку 0,5 н. розсином НCI, потім 1 н. розчином NaOH.
Розчин в двох інших колбах має бути використаний для визначення амінокислот формольним титруванням.
Для цього в кожну з них слід внести витрачену при вказаному вище дослідному титруванні (колба № 1) кількість 0,5 н. розчину НС1 і 0,1 н. розчину NaOH, потім додати по 25 см3 35-40%-ого формаліну, заздалегідь нейтралізованого лугом у присутності індикатора фенолфталеїну.
У колби додати по 1 см3 1%-ого розчину фенолфталеїну та відтитровувати 0,1 н. розчином NaOH до ясного рожевого забарвлення (1 см3 0,1 н. розчину відповідає 1,4 мг азоту амінокислот).
Слід мати на увазі, що разом з амінокислотами титруються присутні у розчині леткі луги. Тому з кількості азоту, визначеного формольним титруванням, слід відняти відповідну кількість летких лугів азоту(АЛЛ). Вміст азоту амінокислот X(у мг/100 г) розраховують за формулою
Х
=
де V - об'єм 0,1 н. розчину NaOH, витрачений на титрування, см3; V1- об'єм 0,1 н. розчину НСl, витрачений на зворотне титрування, cм3; V2 - об'єм рідини в мірному циліндрі, см3; V3 - об'єм фільтрату, взятий для титрування, см3; 1,4 - кількість азоту аміну, що відповідає 1 см3 0,1 н. розчину NaOH, мг; m - маса наважки досліджуваного продукту, г.
Визначення вмісту летких лугів азоту(АЛЛ). До летких лугів (ЛЛ) відноситься ряд з'єднань, у тому числі: аміак NH3, монометиламіни CH3NH2, диметиламіни (CH3)2NH та триметиламіни (CH3)3N.
Кількісний вміст летких лугів азоту є одним з об'єктивних показників свіжості сировини та готової продукції.
Згідно методу пов'язані та вільні леткі луги відгоняються гострою парою. Аміак, що утворився, взаємодіє із сірчаною кислотою. Надлишок кислоти відтитровується лугом.
Підготовка до аналізу та техніка його проведення описані в ГОСТ 7636(п. 3.2.1).
При масових аналізах визначення АЛЛ доцільно проводити на к’єльтек-автоаналізаторі.
Визначення ВУЗ, аміаку та сірководню. Сутність методів, підготовка до аналізу та техніка проведення визначення водоутримуючої здатності м'яса риби, аміаку та сірководню (якісні реакції) описані в ГОСТ 7636 (пп. 3.9; 3.2.3; 3.2.4).
Визначення триметиламіну (CH3)3N титриметричним методом. Азот триметиламіну (ТМА) визначають за різницею вмісту азоту усіх летких лугів (ЛЛ) та азоту аміаку та первинних амінів, якщо в дистиляті були відсутні вторинні аміни. Визначення останніх засновано на здатності їх солей реагувати із формальдегідом (Н2С=О)1 із звільненням еквівалентної кількості кислоти, яку відтитровують лугом.
1 35-40%-ий водний розчин формальдегіду називається формаліном.
Якщо в реакцію вступає амонійна сіль, то звільняється еквівалентна кількість кислоти згідно з рівнянням
6
Н2С=О + 4NH4Сl (H2С)6N4 + 6НО + 4НСl.
Формальдегід Уротропин
Сольові з’єднання первинних амінів реагують подібно хлориду амонію
C
H3NH2HCI + Н2С = О CH3NCH2 + Н2О + НСI.
Отже, одночасно з аміаком формольним титруванням будуть визначатися і первинні аміни, якщо вони знаходяться у розчині.
Сольові з'єднання вторинних амінів реагують із формаліном без відщеплення вільної кислоти, тому присутність їх не заважає визначенню аміаку:
2
(C2H5)2NHHCl + Н2С = О (С2Н5) N2СН2НСl + Н2О.
Третинні аміни не реагують із формальдегідом.
Проведення аналізу. Пробу досліджуваного зразку масою 100 г, відважену з похибкою 0,1 г, переносять з 50 см3 дистильованої води у відгонну колбу місткістю 1000 см3, додають 1 г оксиду магнію та для уникннення спінювання шматочок чистого парафіну.
Перегонку проводять без пари впродовж 1 год, вважаючи з моменту закипання вмісту колби. Дистилят збирають у конічну колбу, що містить 30-50 см3(0,05 міль/дм3) 0,1 н. розчину сірчаної кислоти, в яку має бути занурений кінець трубки холодильника.
Після закінчення перегонки кінець трубки холодильника обмивають дистильованою водою у приймальну колбу та надлишок сірчаної кислоти в приймачі відтитровують0,1 н. розчином їдкого натру з індикатором метиловим червоним (стандартний дослід).
За результатами титрування судять про кількість усіх летких лугів у наважці досліджуваного зразка.
Паралельно з робочим дослідом проводять контрольний дослід (без наважки досліджуваного зразка, але з дотриманням усіх інших умов).
До відтитрованої рідини додають 10 крапель суміші індикаторів бромтимолблау – фенолрот (по 0,2 г кожного індикатора розчиняють у 100 см3 60%-ого спирту) та 20 см3 формаліну, заздалегідь нейтралізованого 0,1 н. розчином їдкого натру у присутності того ж індикатору. Розчин набуває жовто-зеленого забарвлення. Кислоту, що виділилася, відтитровують 0,1 н. розчином лугу до переходу забарвлення розчину від жовто-зеленої до фіолетової.
Вміст азоту триметиламіну X(у мг/100 г) у досліджуваному зразку розраховують за формулою:
Х
=
де V - об’єм 0,1 н. розчину NaOH, витрачений на титрування надлишку H2SO4 у контрольному досліді, см3; V1 - об'єм 0,1 н. розчину NaOH, витрачений на титрування надлишку H2SO4 у стандартному досліді, см3; V2 - об'єм 0,1 н. розчину NaOH, витрачений на титрування розчину після додавання нейтрального формаліну, см3; 1,4 - кількість азоту, еквівалентна 1 см3 0,1 н. розчину NaOH, мг; К - коефіцієнт перерахунку на точно 0,1 н. розчин NaOH; m - маса зразка, узята на аналіз, г.
При визначенні ТМА у відтитрованому розчині, що залишився після визначення АЛЛ, його необхідно кип'ятити упродовж 10-15 хв для видалення діоксиду вуглецю, охолодити спочатку на повітрі, а потім на льоду, заздалегідь закривши колбу резиновою пробкою зі вставленою в неї трубкою, наповненою натровим вапном (сумішшю гідроксиду натрію з гідроксидом каль-цію).
До охолодженого розчину додають 10 крапель суміші індикаторів (бромтимолблау - фенолрот), 20 см3 формаліну, нейтралізованого 0,1 н. розчином NaOH, титрують 0,1 н. розчином лугу, як описано вище. Зміст ТМА розраховують за формулою, приведеній раніше.
Визначення триметиламіну методом спектрофотометрії. Метод заснований на взаємодії амінів з пікриновою кислотою, утворенні забарвленого комплексу та кількісному визначенні триметиламіну спектрофотометричним методом.
Проведення аналізу. До 25 г фаршу риби, відважених з похибкою не більше 0,1 г, у конічну колбу місткістю 200 см3 з притертой пробкою або у приймач автоматичної мішалки підливають 75 см3 5%-ого розчину трихлороцтової кислоти. Суміш енергійно перемішують упродовж 2 хв, після чого наполягають впродовж 1 год при періодичному взбовтуванні. Потім вміст профільтровують через сухий складчастий фільтр, відбирають піпеткою 20 см3 фільтрату у склянку із притертою пробкою, додають 5 см3 20%ого розчину формаліну та перемішують. При необхідності процес може бути перерваний на цій стадії та проба може зберігатися визначений час при 0 0С.
Залежно від передбачуваної концентрації ТМА відбирають піпеткою 0,2-4,0 см3 екстракту у пробірку з поліетиленовою пробкою та доводять об'єм до 4 см3 додаванням дистильованої води (контрольна проба).
У кожну пробірку додають 1 см3 20%-ого розчину формаліну, 10 см3 промитого толуолу С6Н5СНз та 3 см3 насиченого розчину карбонату калію (К2СО3). Закривають пробірку поліетиленовою пробкою, струшують 80 разів, дають вміщеному відстоятися 10 хв та обережно відбирають 8-9 см3 толуолу в іншу пробірку, що містить 100 мг сухого сульфату натрію. Пробірку закривають сухою поліетиленовою пробкою та добре струшують. Акуратно піпеткою переносять 5 см3 сухого толуолу в іншу пробірку, додають 5 см3 0,02%-ого розчину пікринової кислоти [2,4,6-тринитрофенол – С6Н2ОН(NО2)3] та перемішують (робоча проба). Розчин переносять у кювету шириною 10 мм та вимірюють оптичну щільність на спектрофотометрі при довжині хвилі 410 нм по відношенню до контрольної проби.
За градуювальним графіком знаходять концентрацію азоту триметиламіну у розчині, взятому на проведення кольорової реакції, а потім перераховують на її вміст у м'ясі риби.
Вміст азоту триметиламіну в 100 г м'яса (фаршу) риби X (у мг/100 г) розраховують за формулою:
Х
=
де С- вміст азоту триметиламінуу в об'ємі розчину, узятому на проведення кольорової реакції, визначене за градуювальним графіком, мг; V - об'єм витягу, см3; V1 - об'єм фільтрату, взятий до додавання формаліну, см3 (20 см3); V2 - об'єм розчину після додавання формаліну, см3 (25 см3); V3 - об'єм розчину, взятий на проведення кольорової реакції, см3 (5см3); m - маса проби досліджуваного зразку, г.
Побудова градуювального графіку. Для побудови градуювального графіку у ряд пробірок із поліетиленовими пробками вносять 0,5-3 см3 стандартного солянокислого розчину триметиламіну [(СНз)зNНСl] та дистильованою водою доводять об'єм до 4 см3. У одну із пробірок підливають 4 см3 дистильованої води (контрольна проба).
З приготованими розчинами солянокислого триметиламіну різної концентрації проводять такі ж операції, як і з випробовуваним екстрактом, вимірюють оптичну щільність забарвлених розчинів по відношенню до контрольної проби.
За отриманими даними будують градуювальний графік залежності оптичної щільності від концентрації триметиламіну, відкладаючи на осі абсцис концентрацію азоту триметиламіну (у мг) в об'ємі стандартного розчину, взятого у пробірку для визначення, а на осі ординат - величину оптичної щільності забарвлених розчинів.
Концентрація азоту триметиламіну в стандартному розчині має бути виражена відповідно до даних, отриманих при визначенні азоту у стандартному розчині триметиламіну.
Приготування розчинів. 1. Основний розчин солянокислого триметиламіну. 682 мг солянокислого ТМА розчиняють у 100 см3 дистильованої води, що містить 1 см3 концентрованої соляної кислоти (d = 1180 кг/м3). 1 см3 розчину містить близько 1 мг азоту (має бути перевірений мікрометодом).
2. Стандартний розчин. 1 см3 основного розчину додають в 100 см3 дистильованої води, що містить 1 см3 концентрованої НСl.
3. Розчин формаліну 20% - вий. 40% - вий розчин формаліну збовтують з карбонатом магнію до знебарвлення та фільтрують. Фільтрат переганяють та відбирають фракцію зі температурою кипіння 96-98 °С. Перегнаний формалін розчиняють у рівній кількості (об'ємі) дистильованої води.
4. Розчин пікринової кислоти 0,2% - вий. Невелику кількість кислоти висушують в ексикаторі впродовж декількох тижнів. Навішування сухої пікринової кислоти 0,02 г розчиняють в мірній колбі місткістю 100 см3 в сухому толуолі.
5. Сухий толуол. Толуол струшують в ділильній воронці з розчином H2SO4(28 см3 концентрованої кислоти в 100 см3 дистильованої води) в співвідношенні 5:1. Суміш розділяють. Промитий толуол переганяють та обробляють сухим сульфатом натрію.
Реологічні показники. Реологічні1(структурно-механічні) показники гідробіонтів (еластичність, пружність, клейкість, пластичність, в'язкість) відіграють важливу роль в оцінці їх якості.
1 Реологія - наука про деформацію та течію дисперсних систем.
Академік П. А. Ребіндер надавав велике значення використанню реологічних характеристик сировини та продукції в рішенні теоретичних характеристик та практичних завдань. Без знання реологічних показників харчових дисперсних систем2 неможливе не лише вивчення закономірностей їх утворення, але і, що особливо важливо, обґрунтування оптимальних параметрів технологічних процесів.
2 Тканини гідробіонтів є висококонцентрованими дисперсними системами.
Реологічні показники, визначені інструментальними методами, об'єктивно характеризують стан досліджуваної сировини або продукту. Вони добре корелюють з органолептичними та фізико-хімічними показниками.
Тканини гідробіонтів володіють як пружними, так і пластичними властивостями. Тому часто виникає необхідність визначення як оборотних пружно-еластичних, так і безоборотних пластичних деформації.
Деформацією називають відносне зміщення часток, при якому не порушується безперервність самого тіла. Здатність деформуватися під дією зовнішніх сил - основна властивість матеріалів усіх реальних тіл.
Деформації ділять на пружні, тобто зникаючі після зняття навантаження, та залишкові, безоборотні, не зникаючі після видалення навантаження. Залишкові деформації, що не супроводжуються руйнуванням матеріалу, називають пластичними, а самі матеріали – пластичними.
Велике значення при визначенні вказаних деформацій мають температура та швидкість деформації. Щоб змінити форму або деформувати тіло, необхідно прикласти до нього силу Р, зосереджену на визначеній площі та перебільшуючу внутрішній опір його структури. Таку зосереджену силу, віднесену до одиниці площі, називають напрогою3. Воно має певну розмірність.
В залежності від напряму дії сили Р отримують напруги зсуву, стискання та розтягування.
При дослідженні складних багатофазних та гетерогених4 харчових продуктів, в тому числі структур гідробіонтів, частіше визначають деформацію зсуву.
4 Система, що складається з двох або декількох роздільних фаз, тобто однорідних частин, що розрізняються за властивостями або фізичною будовою.
Зсув, як деформація усебічного стискування, є простою деформацією. Усі інші види деформації є більш складними похідними зсуву. Основна трудність при вимірюванні деформації розтягування та стискування - безперервна зміна в процесі виміру площі поперекового перерізу випробовуваного зразка.
Якщо продукт має невисоку пружність та в'язкість, практично неможливо сформувати з нього зразок правильної форми та закріпити його в затисках вимірювального прибору, працюючого на розтягування або стискання. Крім того, опір тіла зсуву в 2,4-3 рази менше, ніж його опір розтягуванню або стисканню, а отже, вимірювання деформації зсуву дозволяє оцінити структурно-механічні властивості продукту з більшою точністю.
За допомогою деформації розтягування, стискання та вигину оцінють пружні та найміцніші властивості сировини і продуктів твердого агрегатного стану.
Для визначення напруги розтягування і стискання необхідно знати розміри досліджуваного зразку і силу, що діє на площу його поперекового перерізу. Діленням величини сили на розмір площі визначають діючу робочу напругу U в грамах на квадратний сантиметр.
Визначення величини граничної напруги зсуву (зсувної міцності) фаршу та готових виробів. Якість фаршу та готових виробів краще всього характеризує величина граничної напруги зсуву. В порівнянні з пластичною і ефективною в'язкістю гранична напруга найбільш чуттєво до зміни технологічних і механічних чинників.
Визначення проводять за допомогою конічних пластомірів з різними видами інденторів (конус, куля, голка та ін.).
Нині для визначення граничної напруги зсуву в різних галузях харчової промисловості використовують пенетрометр-автомат ПМДП, розроблений Московським державним університетом прикладної біотехнології (мал. 12).

Мал. 12. Принципова схема автоматичного пенетрометра ПМДП :
1 - індентор; 2 - панель столу; 3 - контейнер з продуктом; 4 - електромагнітне гальмо; 5 - індикатор переміщення механізму; 6 - електродвигун; 7-схема управління механізмів; 8 - задатчик часу; 9 - лічильник; 10 - цифровий індикатор; 11- перфорований диск; 12 - руків'я переміщення столу
Підготовка проби до вимірювання. Філе риби обезшкірюють, видаляють кістки та подрібнюють на м'ясорубці, що має решітку з отворами діаметром 3 мм. Фарш за допомогою шпателя поміщають у циліндричну склянку, вирівнюючи (шпателем, ножем) його поверхню1.
1 Морожений фарш розморожують на повітрі при кімнатній температурі до 2...5 °С в товщі брикету, двічі подрібнюють на м'ясорубці, нагрівають у склянці на водяній бані при температурі 30 0C до 18...20 0C, після чого укладають у циліндричний металічний стакан.
При наповненні склянки фаршем допускається періодичне постукування його по столу з метою видалення повітря та рівномірного заповнення фаршем. Після наповнення фарш підпресовують при мінімальному навантаженні (не більше 0,16 кгс/см2= 15690,6 Па; при роботі з мороженим фаршем - 0,04 кгс/см2 = 3700 Па). Заздалегідь поверхню банки накривають поліетиленовим гуртком. Тривалість підпресовування 5 хв.
Для зняття внутрішньої напруги після підпресовування зразок витримують при кімнатній температурі впродовж 5 хв. Для прискорення підсихання поверхневого шару фаршу склянку із зразком закривають кришкою.
Проведення вимірювання. Циліндричну склянку з досліджуваним зразком фаршу поміщають на стіл пенетрометра під вістря індентору (конусу) та натискають пускову кнопку пенетрометра, заздалегідь підключеного до джерела електроживлення (мережа). Штанга з індентором вільно падає та індентор занурюється в досліджуваний зразок. Початок моменту вимірювання (занурення, зіткнення) фіксується автоматично. У міру занурення площа зіткнення індентора з масою зростає та швидкість занурення зменшується. Тривалість вимірювання 180 із [подальшим зануренням конусу (голки) на незначну величину можна знехтувати]. У момент визначення граничної напруги зсуву температура зразку в центрі склянки має бути 20±1°С.
Виміри проводять в центрі кожного з трьох приготованих зразків.
Значення
граничної напруги зсуву
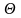 (у Па) розраховують за формулою П. А.
Ребіндера
(у Па) розраховують за формулою П. А.
Ребіндера

де К- константа конусу, Н/кг (для конусу з кутом 10, 20, 30, 45, 60, 90° постійна рівна відповідно 71,05; 20,30; 9,40; 4,08; 2,10; 0,715); m- маса наважки (загальна - маса утримувача, індентора) без тертя, кг; h- глибина занурення конуса, м.
За результат випробувань приймають середнє арифметичне трьох визначень, закруглене до значень, кратних п'яти. Гранична напруга зсуву мороженого рибного фаршу від 500 (добре формується) до 1300 Па (фарш не формується).
Визначення токсичних речовин. З їжею в організм людини поступає 2/3 усіх забруднень. Тому безпеці продуктів, отриманих з гідробіонтів, приділяється велика увага.
Забруднення, що поступають у водойму, за складом та впливом на біосферу можна розділити на дві групи:
органічні речовини, здатні розкладатися (білкові речовини, жири і т. д.);
хімікалії (токсичні хімічні елементи, пестициди і т.д.).
Остання група забруднюючих речовин найбільш багаточисленна та різноманітна за характером дії на біосферу. Багато забруднюючих речовин не використовуються мікроорганізмами і тому не розкладаються і не беруть участь у біологічному кругообігу речовин (наприклад, інсектицид ДДТ та ін.).
Особливу небезпеку викликає вступ токсичних речовин у гідросферу. Дія їх на водні організми різко знижує продуктивність водойм внаслідок загибелі об'єктів, які служать їжею для риб та інших промислових гідробіонтів. Крім того, маючи кумулятивні властивості, деякі токсичні речовини накопичуються в організмі риб, морських ссавців, ракоподібних, молюсків та інших об'єктів промислу (наприклад, ДДТ та його метаболіты - ДДД, ДДЕ; альфа- та гамма-ГХЦГ; поліхлоровані біфеніли (ПХБ)1 та інші речовини.
1 За хімічною номенклатурою – дифеніли.
Усі вивчені пестициди відрізнялися мутагенною активністю, тобто здатністю змінювати спадковість.
За фізико-хімічними властивостями та фізіологічному впливу на організм дуже близькі до хлорорганічних пестицидам поліхлорбіфенілы (ПХБ). Хоча загальна токсичність їх невелика (більше 400 мг/кг), хронічна токсичність представляє серйозну небезпеку для людини (хвороба "Юхо" - висип, свербіж, потемніння шкіри, розлад зору).
Встановлена висока стабільність біфенілів в морській воді, яка набагато вище стабільності ДДТ та його метаболітів.
Чималу небезпеку представляє кумулювання у гідробіонтах токсичних хімічних елементів(ртуть, свинець та ін.).
Ртуть - одно з найбільш небезпечних металевих забруднень. Основна частина їх представлена метилртуттю(CH3Hg+ ), що проникає в кров та з’єднується з білками плазми. Велика частина метилртуті концентрується в еритроцитах. Більше 90 % ртуті в тканинах мозку знаходиться в органічній формі (метилртуть, етилртуть).
При підвищеному вмісті ртуті в організмі виникає хвороба "Минамата" (погіршення зору, оніміння рук та ніг, невиразна мова).
Патологія частин тіла, органів та крові спостерігається і при підвищеному вмісті в них свинцю.
Типовим проявом отруєння кадмієм є пошкодження нирок (хвороба "Ітай-Ітай"). Пошкоджуються проксимальні канальці нирок, чому порушується їх адсорбційна функція. Після припинення дії кадмію процеси адсорбції не відновлюються.
Здавна вважається отрутою миш'як. Гідробіонти (риби, ракоподібні та ін.) акумулюють цей елемент. Найбільш токсичні з'єднання тривалентного миш'яку (оксид миш'яку Аs2О3).
Миш'як - отрута, що діє на протоплазму клітини. Взаємодіючи з сульфгідрильними групами (-SH-), він інгібує дію ферментів, особливо тих, які беруть участь в процесі клітинного метаболізму(обмін речовин) та дихання.
Хронічне отруєння призводить до втрати апетиту, зниженню маси тіла, гостро кишковому розладу, меланоми шкіри та інших негативних наслідків.
Негативну дію на життєдіяльність організму людини чинить також підвищений вміст в продуктах та інших токсичних елементів (наприклад, цинку, хрому, міді, алюмінію), а також нітратів, нітриту, гістаміну, летких N- нітрозамінів та інших токсичних речовин.
Нітрити більш токсичні за нітрати. Взаємодіючи з гемоглобіном крові, нітрати переводять його двовалентне залізо у тривалентне, властиве метгемоглобіну, позбавляючи його здатності транспортувати кисень, і тим самим порушують нормальне тканинне дихання.
Відомо, що такі токсичні речовини, як гістамін та N- нітрозаміни, можуть утворюватися в сировині і продуктах в процесі їх виготовлення.
Гістамін (C5N3H9) - біогенний амін - відноситься до біотоксинів. Він знижує кров'яний тиск, послабшує скорочення гладких м'язів, змінює проникність кровоносних судин, порушує діяльність підшлункової залози та викликає алергію.
Природний вміст гістаміну у тканинах організму тварини невеликий, і тому він не чинить несприятливої дії на організм.
У сировині та продуктах гістамін утворюється в результаті декар-боксилювання амінокислоти - гістидину - за участю фементів мікрофлори.
У перетворенні гістидину на гістамін беруть участь бактерії групи кишкової палички, бактерії роду Proteus (Proteus morgani - найбільш активний продуцент гістаміну), споротворні анаеробні бактерії роду Clostridium (Salmonella), а також деякі види молочнокислих бактерій (Lactobacіllus), що розвиваються при недотриманні режимів технологічних процесів та умов зберігання сировини і продуктів.
Накопичення гістаміну в гідробіонтах може відбуватися в період від вилову до заморожування, причому особливо інтенсивно, якщо зберігати їх без охолодження.
Можливе накопичення гістаміну в рибі і в результаті порушення ланцюга холодильного зберігання, недотримання технологічного режиму процесу розморожування та строків зберігання сировини та напівфабрикатів перед термічною обробкою.
Гістамінові отруєння найчастіше спостерігаються при використанні продуктів переробки скумбрієвих.
Прихований період отруєння рибою з підвищеним вмістом гістаміну зазвичай складає менше 1 год, проте в залежності від індивідуальних особливостей організму може коливатися від 5 хв до 5 год.
Симптоми гістамінової інтоксикації характерні. Постраждалі відмічають різкий або гіркий (перцевий) смак їжі. Спостерігаються почервоніння лиця і шиї з почуттям жару та загального дискомфорту, сильні головні болі, сльозотеча, болі в очах, набряк слизової оболонки носа. Іноді спостерігаються порушення серцевого ритму, паління в порожнині рота та глотки, утруднення ковтання, поява висипу на обличчі та шиї, свербіж шкірних покривів. У деяких постраждалих відзначаються шлунковий розлад, болі в животі, рідке випорожнення. У важких випадках спостерігаються шок, бронхоспазми, задуха та розлад дихання.
Потенційно небезпечними для здоров'я людини речовинами, виявленими в продуктах харчування, є N- нітрозаміни. З 120 вивчених нітрозоз’єднань 80 % відносяться до канцерогених (N- нітрозодиметиламін, N- нітрозодиетиламін, N- нітрозодипропіламін, N- нітрозопиперидин та ін.). Навіть низькі концентрації нітрозаміну в короткі терміни викликають у тварин злоякісні пухлини і, поза сумнівом, небезпечні для людини.
До попередників нітрозоз’єднань відносяться, з одного боку, нітрати, нітрити та оксиди азоту, з іншої – аміни (вторинні, третинні) та аміди.
Аміни та аміди, будучи проміжними продуктами в метаболізмі білкових речовин, також постійно є присутніми в харчових продуктах. У значних кількостях вони містяться в рибі, м'ясі наземних тварин та інших продуктах.
Аміни - органічні сполуки, утворені в результаті заміщення атома водню у вуглеводній молекулі залишками аміаку (аміногрупами : первинні аміни R - NH2; вторинні аміни R - NH – R1; третинні аміни R - N – R3, де R, R1 - вуглеводневі радикали (-СН3, -С2Н5,...).
Вивчені умови, при яких аміни, аміды та амінокислоти можуть вступати у взаємодію з нітратами за допомогою нітрозування (заміщення водню в органічних з’єднаннях нітрозогрупою з утворенням нітрозоз’єднань).
Наявність N- нітрозамінів в продуктах, що виробляються з гідробіонтів, обумовлена в основному освітою їх в період консервації (обробки) сировини речовинами, з’єднаними нітрити (хлорид натрію, коптильний дим та ін.), а також стерилізацією, висушуванням і в'яленням риби та інших об’єктів.
Можливість багатьох токсичних речовин накопичуватися у гід-робіонтах та утворюватися в продуктах, що виробляються з них, призводить до необхідності проведення постійного контролю за рівнем їх вмісту в сировині та продукції.
Нині в гідробіонтах та продуктах, що виробляють з них, передбачено визначення токсичних хімічних елементів (загальна ртуть, свинець, кадмій, миш'як, мідь, цинк, олово, хром), хлорорганічних пестицидів (ДДТ та його метаболітів – ДДД, ДДЕ; альфа- та гамма-ГХЦГ, гексахлорану), 2,4-дихлорфеноксиоцтової кислоти (2,4-Д), поліхлорованих біфенілів (ПХБ), гістаміну, N- нітрозамінів, 3,4-бенз[а]пірену, діоксинів - у перерахунку на 2, 3, 7, 8-тетрахлордибензо[b, е]-1,4-диоксин(2, 3, 7, 8-ТХДД), радіонуклідів - цезію- 134, цезію- 137; стронцію- 90.
Визначення загальної ртуті методом безполумневої атомної абсорбції. Сутність методу, підготовка до аналізу та техніка проведення його описані в ГОСТ 26927 і ГОСТ 7636(п. 3.8).
Визначення свинцю, кадмію, миш'яку, міді, цинку, олова, хрому та заліза методом атомної абсорбції 1. Метод заснований на полум'яній або безполумневій атомній абсорбції. Атоми хімічних елементів здатні збуджуватися під дією світової енергії, поглинаючи світло відповідних частот, які співпадають із частотами випромінювання атомів цих елементів.
1 Визначення елементів проводиться також стандартними методами: миш'яку - за ГОСТ 26930, міді - за ГОСТ 26931, свинцю - за ГОСТ 26932, кадмію - за ГОСТ 26933, цинку - за ГОСТ 26934, олова - за ГОСТ 26935.
Поглинання світової енергії прямо пропорційна концентрації елементу, присутнього в атомній хмарі.
Для атомизації проби використовують теплову енергію полум’я або електричну енергію, застосовуючи графітові кювети.
Мінералізацію проби проводять сухим або мокрим способом. При визначенні миш'яку мінералізацію проби проводять тільки сухим способом (озолення) з додаванням нітрату магнію.
Відбір і підготовка проб. Проби риби, морських ссавців, безхребетних та продуктів, що виробляються з них, відбирають за ГОСТ 7631, проби водоростей - за ГОСТ 20438, консервів та пресервів - за ГОСТ 8756.
Проби готують до аналізу у відповідності до ГОСТ 7636, ГОСТ 16280, ГОСТ 1720, ГОСТ 22455, ГОСТ 6730, ГОСТ 20438, ГОСТ 26185.
Проведення аналізу. 1. Суха мінералізація. Наважку досліджуваного зразку, подрібненого за допомогою скальпелю, ножиць або шляхом розтирання у фарфоровій ступці при досліді консервів (використання м'ясорубки не допускається), масою 5...40 г (кінцева маса золи має бути 0,4-0,5 г), узяту з похибкою не більше 0,01 г, поміщають у кварцевий тигель та висушують при температурі 105 °С; потім обережно обуглюють та озолюють у муфельній печі, повільно підвищуючи температуру від 130 до 450 °С. Піднімають температуру до 250-270 °С поступово: на 20-30 °С, далі на 40-50 °С; при цьому необхідно уникати "димлення", оскільки з димом можливі втрати металів. Зразок витримують при температурі 450 °С до отримання однорідно забарвленої золи світлого кольору. Якщо отримана зола містить темні вкраплення, після охолодження змочують її азотною кислотою (1: 1) та продовжують озолення до отримання світлої золи. До охолодженої золи додають 5-10 см3 соляної кислоти (1:5), нагрівають на електроплитці до повного розчинення, переносять отриманий розчин в мірну колбу місткістю 25 см3 через обеззолений фільтр та доводять об'єм дистильованою водою до мітки.
2. Суха мінералізація для визначення миш'яку. Наважку гомогенізованого зразку (1-4 г для вологої рибної тканинита 0,5 г для висушеної тканини), взяте з похибкою не більше 0,01 г, поміщають у кварцевий тигель місткістю 50-100 см3, додають 8 см3 водного розчину нітрату магнію (500 г/дм3) та ретельно перемішують із зразком. Зразок висушують при температурі 105 °С, потім обережно обуглюють та озолюють в муфельній печі, повільно підвищуючи температуру до 450 °С, уникаючи "димлення". Витримують зразок при 450 °С впродовж 12 год. Після охолодження розчиняють золу в 10 см3 концентрованої соляної кислоти та доводять об’єм дистильованою водою до 20 см3. Отриманий розчин використовують для визначення миш’яку за допомогою миш’якового аналізатору.
3. Мокра мінералізація. Наважка досліджуваного зразку, виготовленого, як описано вище, масою 1,5-2,0 г, взяту з похибкою не більше 0,001 г, поміщають у заздалегідь висушену до постійної маси широкогорлу колбу місткісю 50-100 см3 та висушують до постійної маси у сушильному шафі при температурі 100-105 °С. Висушену наважку заливають 5 см3 концентрованої азотної кислоти, закривають пробкою та залишають на ніч. Азотну кислоту в випаровують з колби до вологого стану наважки, потім тричі додають по 10 см3 дистильованої води, кожного разу випаровуючи до вологого стану зразка.
Вміст колби переносять дистильованою водою через фільтр в мірну колбу місткістю 25 см3, об'єм рідини доводять до мітки, ретельно перемішують та фотометрують при певній довжині хвилі.
Розчин мінералізата розпилюють у легко-ацетиленовому полум'ї. Величину резонансного поглинання атомів визначеного елементу вимірюють за допомогою атомно-абсорбційного спектрофотометра.
Кількість металу, що відповідає певній висоті сигналу, знаходять по градуювальному графіку.
Обробка результатів. Масову частку досліджуваного елементу (у млн.-1) визначають за формулою
Х
=
де m1 - вміст досліджуваного металу, знайдений за градуювальним графіку, мкг/см3, m2 - маса досліджуваного зразку, г; V - об'єм, до якого доводять дистильованою водою досліджувану пробу, см3.
За остаточний результат приймають середнє арифметичне значення результатів двох паралельних визначень.
Визначення хлорорганічних пестицидів (ДЦТ та його метаболітів - ДДД, ДДЕ; альфа- та гамма-ГХЦГ) газохроматографічним методом. Метод заснований на витяганні пестицидів з проби органічним розчинником або сумішшю розчинників, очищенні екстракту та визначенні пестицидів газової хроматографії.
Відбір та підготовка проб. Проби риби, морських ссавців, безхребетних та продуктів з них відбирають за ГОСТ 7631, водоростей - за ГОСТ 20438, муки - за ГОСТ 13496, консервів та пресервів - за ГОСТ 8756. 0.
При спеціальних дослідженнях відбір проб проводять у відповідності з методичними вказівками Міністерства охорони здоров'я СССР № 2051-79, затвердженими 25 серпня 1979 р.
Рибу очищають від луски, видаляють внутрішні органи та розбирають на філе. Досліджуваний зразок (філе1 та ін.) масою близько 250 г подрібнюють 1-3 рази на м'ясорубці (в залежності від консистенції тканини) з діаметром отворів решіток 3 мм та ретельно перемішують. Для більш тонкого подрібнення зразку (окрім жирової тканини) фарш рекомендується додатково подрібнювати у гомогенізаторі.
1 Наявність шкіри вказують.
Пробу печінки морських ссавців масою 100-200 г подрібнюють на м'ясорубці, ретельно перемішують і у разі потреби додатково подрібнюють у гомогенізаторі. Жирну печінку риб (наприклад, тріски) заздалегідь не подрібнюють.
Пробу жиру розігрівають до температури, передбаченої стандартом при визначенні прозорості (40-50 °С) в залежності від виду жиру, ретельно перемішують та фільтрують через паперовий фільтр в чисту суху пробірку (колбу) з притертою пробкою.
Витягання пестицидів з наважки досліджуваного зразку проводять гексаном або сумішшю розчинників - гексану та ацетону.
Наважку досліджуваного зразку масою 10 г, зважене з похибкою не більше 0,01 г, розтирають частинами у фарфоровій ступці із безводним сульфатом натрію (у співвідношенні 1: 4) до отримання розсипчастої маси та переносять її в плоскодонну колбу з притертою пробкою місткістю 250 см3. У колбу вносять 60 см3 ацетону, 60 см3 гексану та екстрагують ліпіди впродовж 2 год, перемішуючи вміст колби в апараті для струшування. Після цього суміш фільтрують на воронці Бюхнера через обеззолений паперовий фільтр з червоною смугою. Осад на фільтрі двічі промивають гексаном(15 і 10 см3). Отриманий екстракт переносять в ділильну воронку, обережно струшують вміст та дають суміші відстоятися. Після відстоювання нижній ацетоновий шар зливають в колбу. Шар гексану профільтровують через шар безводного сульфату натрію (30-40 г). З ацетонового шару пестициди двічі екстрагують гексаном (15 та 10 см3). Отримані екстракти фільтрують через той же сульфат натрію.
Екстракти гексанів об'єднують, концентрують на роторному випарнику при температурі 35 °С до 4 см3 та переносять в ділильну воронку. У воронку підливають концентровану сірчану кислоту(приблизно у співвідношенні 1: 6 за об'ємом) та обережно струшують, не допускаючи утворення емульсії на межі розділу суміші.
Кислотну фазу відкидають та додають нову партію сірчаної кислоти. Очищення продовжують до тих пір, поки кислота не перестане забарвлюватися та каламутніти. Після цього екстракт гексану промивають дистильованою водою до нейтральної реакції (по лакмусовому папірцю) та профільтровують через воронку з безводним сульфатом натрію та отриманий об'єм екстракту гексану доводять до 5 см3.
З пробірки мікрошприцем, заздалегідь промитим гексаном, відбирають 1 мкл розчину пестицидів та вводять його у газовий хроматограф, використовуючи капілярну колонку та детектор електронного захоплення (ЕСД), заздалегідь підготовлений до роботи.
При проведенні аналізу необхідно строго дотримуватися класифікації використовуємих реактивів.
Чистоту гексану та ацетону перевіряють на газовому хроматографі.
Для цього 100 см3 розчинника упарюють до 5 см3, відбирають
для аналізу 1 мкл розчинника та вводять його у хроматограф. Поява на хроматограмі додаткового піку свідчить про недостатню чистоту розчинника.
Очищення розчинників здійснюють перегонкою при температурі 56-60 °С (для ацетону) та 68,7-70 0С (для н - гексану).
При виконанні роботи необхідно строго дотримуватися правила по техніці безпеки.
Техніка відділення хлорорганічних пестицидів в рибі та рибних продуктів хроматографічним методом з використанням набивної колонки описана в тимчасових методичних вказівках, затверджених Міністерсвом охорони здоров'я СССР 22 жовтня 1981 р. № 2482 – 81.
Визначення гістаміну. Метод заснований на кількісному фотометричному визначенні величини абсорбції забарвленого продукту взаємодії гістаміну з диазореактивом.
Відбір та підготовка проб. Відбирають та готують проби згідно ГОСТ 7631, ГОСТ 7636, ГОСТ 8756 та діючій нормативній документації на конкретний вид продукції. Зразки доставляють в лабораторію відразу після відбору проби. При тривалому транспортуванні (більше 1 год) рибу-сирець необхідно транспортувати в охолодженому стані, морожену - в замороженому стані. Зразки досліджують в день доставки їх в лабораторію. Інакше зразки зберігають при температурі не вище мінус 8 °С упродовж не більш 3 діб.
Доставляють та зберігають зразки консервів та іншої продукції з дотриманням режимів, передбачених діючими інструкціями та стандартами.
Апаратура, реактиви, матеріали. Ваги лабораторні загального призначення з метрологічними характеристиками за ГОСТ 24101 з найбільшою межею зважування 200 г, 2-го класу точності.
Ваги лабораторні загального призначення з метрологічними характеристиками за ГОСТ 24104 з найбільшою межею зважування 1 кг, 4-го класу точності.
Воронка ділильна ВД- 3-250-2932 ХС по ГОСТ 25336.
Воронки В- 100-150 ХС; В- 35-50 ХС по ГОСТ 25 336.
Колба конічна 1-100-29/32 МС по ГОСТ 25336.
Колба мірна 1-100-2 або 2-100-2 по ГОСТ 1770.
Колориметр
фотоелектричний за ГОСТ 12083 зі світофільтром
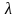 =
490±10 нм, спектрофотометр СФ- 26 або
аналогічний.
=
490±10 нм, спектрофотометр СФ- 26 або
аналогічний.
Мікроподрібнювач тканин РТ- 2 або аналогічний.
Піпетка 4-1-1 або 5-1-1; 7-1-5 або 7-2-5; 7-1-10 або 7-2-10 за ГОСТ 20292.
Пробірка мірна П- 2-10 або П- 2-15 за ГОСТ 1770.
Термометр 0...100 °С за ГОСТ 2045.
Холодильник побутовий.
Шафа лабораторна сушильна.
Циліндр мірний 1-50 або 2-50 за ГОСТ 1770.
Електроплитка битова за ГОСТ 14919.
Папір фільтрувальний за ГОСТ 12026.
н-Бутанол за ГОСТ 6006, ч.
Вода дистильована за ГОСТ 6709.
Гистамін (C5H9N3) або гідрохлорид (C5H9N3 • 2НС1).
Кислота соляна за ГОСТ 3118, х. ч.; розчин 3,75 г/дм3 (0,1 н. розчин).
Кислота трихлороцтова за ТУ 6-09-1926, розчин 50 г/дм3 (5%-вий розчин).
Натрій азотистокислий (нітрат натрію) за ГОСТ 4197, ч. д. а., розчин 50 г/дм3 (5%-вий розчин).
Гідроксид натрію за ГОСТ 4328, ч. д. а., розчин 200 г/дм3 (5 н. розчин).
Натрій сернокислий безводний (сульфат натрію) за ГОСТ 4166, х. ч.
Карбонат натрію за ГОСТ 8379, х.ч., розчин 40 г/см3 (4%-вий розчин).
Етилацетат за ГОСТ 22300, ч. д. а.
Підготовка до аналізу. Приготування розчинів. 1. Розчин n-нітроаніліну. n -Нітроанілін очищають шляхом перекристалізації з води. Його розчиняють у гарячій дистильованій воді та отриманий розчин охолоджують до кімнатної температури. Кристали, що випали, фільтрують та висушують при 80 °С упродовж 10-15 хв. 0,1 г n-нітроаніліну розчиняють в 100 см3 0,1 н. соляної кислоти. Зберігають розчин у холодильнику не більше року.
2. Диазореактив отримують безпосередньо перед використанням шляхом змішування 10 см3 розчину, що містить 1 г/дм3 n-нітроаніліну в 0,1 н. соляній кислоті, та 1 см3 розчину, що містить 50 г/дм3 азотистокислого натрію. Розчин охолоджує до 0 °С.
3. н-Бутанол, насичений водою (50 см3 н-бутанолу та 20 см3 дистильованої води), струшують в ділильній воронці. Після розділення фаз відділяють верхній шар, що містить насичений водою бутанол.
4. Стандартні розчини гістаміну. а. Основний стандартний розчин, що містить 40 мкг/см3 гістаміну (4,0 мг гістаміну чи 5,31 мг гідрохлориду гістаміну), поміщають у мірну колбу місткістю 100 см3, розбавляють 5%-вим розчином трихлороцтової кислоти та доводять об'єм до мітки. Розчин зберігають в холодильнику не більш одного місяцю.
б. Робочі стандартні розчини гістаміну. Робітники розчину гістаміну з концентраціями 20, 10 і 5 мкг/см3 отримують шляхом розбавлення 5%-вим розчином трихлороцтової кислоти основного розчину відповідно в 2, 4 і 8 разів.
Робочі стандартні розчини готують в день проведення аналізу.
Побудова калібрувальної кривої. 5 см3 кожного стандартного розчину гістаміну з концентраціями 5; 10; 20 і 40 мкг/см3, приготованих по а і б, поміщають в мірні пробірки з притертими пробками, додають 1 см3 20%-вого розчину гідроксиду натрію та вносять в розчин при перемішуванні безводний карбонат натрію до отримання прозорого розчину. Додають 5 см3 н- бутанолу, насиченого водою(п. 3), та енергійно струшують пробірки упродовж 30 с. Після розділення фаз відбирають піпеткою або шприцом 3 см3 верхнього бутанольного шару та переносять в іншу пробірку з притертою пробкою, що містить 3 см3 0,1 н. розчину соляної кислоти. Струшують вміст пробірки впродовж 30 с. Після розділення фаз відбирають 2 см3 нижнього водного шару, переносять в іншу пробірку, додають 2 см3 4%-вого розчину карбонату натрію та витримують упродовж 5 хв при 0 0С. Підливають до охолодженого розчину 2 см3 холодного свіжоприготованого диазореактиву за п. 2, струшують і знову витримують упродовж 5 хв при 0 0С. Додають 4 см3 етилацетату та енергійно струшують упродовж 30 с. Після розділення фаз сухою піпеткою відбирають верхній шар та переносять в пробірку, що містить безводний сульфат натрію (розчин забарвленого похідного гістаміну має бути прозорим). Вимірюють величину абсорбції при 495 нм у кюветі завтовшки 1 см. В якості розчину порівняння використовують етилацетат. На підставі отриманих даних будують калібрувальну криву залежності величини абсорбції від концентрації гістаміну у розчині.
Виділення гістаміну. Наважка у 10 г досліджуваного продукту поміщають у посудину мікроподрібнювача тканин, додають 25 см3 25%-вого розчину трихлороцтової кислоти та перемішують упродовж 5 хв. Отриману суміш переносять у конічну колбу місткістю 100 см3, ополіскують посудину змішувача двічі (по 5-10 см3 розчину) 5%-вим розчином трихлороцтової кислоти та розчини об'єднують. Колбу забезпечують повітряним холодильником і витримують на водяній бані при 60 °С упродовж 15 хв. Охолоджену суміш переносять кількісно у циліндр, доводять до 50 см3 5%-вим розчином трихлороцтової кислоти та фільтрують через складчастий паперовий фільтр (фільтрат 1).
5 см3 фільтрату 1 поміщають у пробірку з притертою пробкою та проводять процедуру, описану вище, починаючи з додавання розчину гідроксиду натрію.
Якщо зразки містять більше 200 мг/кг гістаміну, необхідно розбавити отриманий фільтрат 5%-вим розчином три хлороцтової кислоти.
Для визначення концентрації гістаміну в екстракті використовують калібрувальну криву, описану вище.
Обробка результатів. Масову частку гістаміну в продукті X (у мг/кг) розраховують за формулою
Х
=

де С - концентрація гістаміну, знайдена по калібрувальній кривій, мкг/см3; V - об'єм екстракту, см3; m- наважка зразку продукту, г; Ф - чинник розбавлення.
Ф
=

За остаточний результат приймають середнє арифметичне двох паралельних визначень.
Границя визначення гістаміну - 10 мг/кг. При концентраціях 10-500 мг/кг величина поглинання пропорційна кількістю гістаміну у розчині.
Міра витягання (виявлення) доданого гістаміну 90-95%.
Відносне стандартне відхилення при визначенні гістаміну концентрацій 10-100 мг/кг – 20%, 101-500 мг/кг – 15%.
Вимоги техніки безпеки. Приміщення, в якому проводиться визначення гістаміну, має бути обладнане приточно-витяжною вентиляцією. Роботу необхідно вести у витяжній шафі з використанням індивідуальних засобів захисту.
Визначення N- нітрозамінів флуориметричним методом. Метод заснований на здатності летких N- нітрозамінів (НА) виділятися з подрібненої проби досліджуваного зразка шляхом перегонки з парою або у вакуумі, екстракції хлористим метиленом (метиленхлоридом) НА з водного дистиляту, концентрування екстракту, денітрозування НА бромистим воднем в оцтовій кислоті, алкілірування амінів 8-метокси-5-хинолінсульфонілазиридином (КАЭ), що утворилися, розділення та кількісного визначення утворившихся флуоресціюючих 8-метокси-5-[N-(2-N-диетиламіно)] хинолінсульфонамідних похідних(КАЕ-похідні) у тонкому шарі силікагелю.
Ідентифікацію НА здійснюють порівнянням рухливості в тонкому шарі силікагелю флуоресціюючих КАЕ-похідних із зразку з рухливістю відповідних стандартних похідних: диметиламіну - КАЕ ДMA, диетиламіну - КАЭ ДЕА, дипропіламіну – КАЕ ДПА.
У основі напівкількісного визначення лежить візуальне порівняння інтенсивності флуоресценції плям КАЕ- похідних із зразку з інтенсивністю флуоресценції плям стандартних з'єднань. При необхідності можливо також кількісне визначення шляхом виміру величини флуоресценції КАЕ- похідних. Нижня межа визначення НА 1 мкг/кг продукту.
Відбір і підготовка проби. Відбір проби рибопродукції та підготовку її до аналізу проводять відповідно до ГОСТ 7631 і 7636, а консервів та пресервів - до ГОСТ 8756.0.
Для визначення N- нітрозамінів з середньої проби відбирають зразок масою 100-200 г в широкогорлу склянку з притертою пробкою, який може зберігатися при температурі мінус 8... мінус 10 °С до 3 діб.
Виділення НА можна проводити шляхом перегонки з парою і у вакуумі.
При виділенні НА шляхом перегонки з парою наважку дослідного продукту в 50-100 г, подрібнене у м'ясорубці або гомогенізаторі, поміщають у круглодонну колбу місткістю 50 см3, з’єднану з паровиком та прямим холодильником(мал. 13).
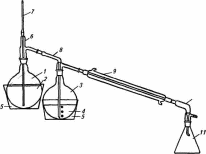
Мал. 13. Схема установки для виділення N- нітрозамінів :
1 - паровик; 2 - дистильована вода; 3 - круглодонна колба; 4 - гомогенат харчового продукту; 5 - баня з гліцерином; 6 - насадка Ворца; 7 – трубка на шліфі; 8 – насадка-барботер; 9 – холодильник Лібіха; 10 - алонж; 11 - приймач
Після додавання різних реактивів проводять відгін НА, збираючи 200-230 см3 дистиляту. Якщо в процесі перегонки відбувається спінювання маси, додають 5-10 см3 насиченого розчину апіезону у ізопропіловому спирті. Отриманий дистилят переносять у ділильну воронку та екстрагують нітрозаміни свіжоприготованим хлористим метиленом (метиленхлоридом) 4-5 разів по 15 см3.
Об'єднані хлорметиленові екстракти висушують прожарюваним сульфатом магнію, фільтрують, осушувач промивають невеликим об'ємом хлористого метилену. Висушені фільтрати поміщають у круглодонну колбу місткістю 100- 150 см3, забезпечують дефлегматором (чи повітряним холодильником), поміщають у водяну баню та упарюють хлористим метиленом (метиленхлоридом) до 5-7 см3 при температурі 55-65 0С. Доцільно пропускати через хлорметиленовий розчин інертний газ.
При досягненні об'єму розчину 5-7 см3 дефлегматор змивають 2 см3 гексану та продовжують концентрувати пробу до 1-2 см3.
При виділенні нітрозамінів перегонкою з вакууму пробу дослідного продукту масою 20-50 г, відважену з похибкою 0,1 г, поміщають у круглодонну колбу місткістю 200 см3 і додають 70-80 см3 дистильованої води. Потім у отриману суміш в якості внутрішнього стандарту додають 1 см3 розчину гексану N- нітрозодипропіламіну (НДПА), 4 см3 0,1 н. розчину гідроксиду натрію і 20 см3 парафінової або вазелінової олії для відвертання перекидання маси під час дистиляції та приєднують до вакуумної системи(мал. 14).

Мал. 14 Схема вакуумної установки для виділення N- нітрозамінів
Колбу з аналізованим продуктом поміщають в піщану лазню, під якою встановлюють газовий пальник. Приймальну колбу охолоджують сумішшю сухого льоду і гексану(ацетону) або рідким азотом. Встановивши в системі вакуум, проводять дистиляцію приблизно 1 год при температурі у дистиляційній колбі 98-100 0С до отримання 50 см3 дистиляту.
Щоб уникнути втрати, бажано проводити дистиляцію в закритій системі, як можна рідше підключаючи її до вакуумного насосу. Герметичність системи можна зберегти, використовуючи вакуумне мастило або тефлонові манжети для шліфтів.
Після закінчення дистиляції припиняють обігрів, охолодження, від'єднують шланг вакуумної системи та закривають накінцівник приймача тефлоном або пробкою.
Після відтавання приймальної колби систему перегонки розбирають, насадку ополіскують декількома мілілітрами дистильованої води, які додають до дистиляту.
У круглодонну колбу з дистилятом додають 1-2 см3 метанолу, 0,5 см3 5 н. сірчаної кислоти та екстрагують тричі по 7-10 см3 хлористим метиленом (метиленхлоридом). Останній збирають піпеткою з дна колби та переносять в скляночку місткістю 50 см3 з 1-2 г безводного сульфату натрію. Після підсушування кожну партію екстракту переносять в пробірку. Сульфат натрію, що залишився в скляночці, промивають 2 см3 хлористого метилену (метиленхлориду), який також переносять у пробірку з екстрактом.
Концентрацію екстракту проводять на водяній бані при температурі 40 0С під струмом азоту до 5-7 см3. Потім екстракт переносять у центрифужну пробірку та концентрують під струмом азоту при кімнатній температурі до 1 см3 та менш (не допускаючи повного відгону екстракту).
Після проведення алкілування (денітрозування гексанового або хлорметиленового розчину та отримання КАЕ- похідних) проводять хроматографування, ідентифікацію нітрозамінівта визначення їх флуориметричним методом (флуориметр ЕФ-ЗМ з фільтрами Bl - 1, В2-1 або інші).
Хемілюмінесцентний метод. Метод ідентифікації та кількісного визначення НА хемілюмінесцетним методом полягає у виділенні летких НА шляхом перегонки з парою або у вакуумі, екстракції хлористим метиленом (метиленхлоридом) НА з водного дистиляту, концентрації екстракту, розділенні суміші методом газорідинної хроматографії та кількісному визначенні немодифікованих НА за допомогою високоселективного та високочутливого хемілюмінесцентного(термоенергетичного) детектора TEA - 502 фірми "Termo Electron corporation"(США). Використовують газовий хроматограф.
Ідентифікацію НА здійснюють за часом утримування, порівнюючи з параметрами утримування стандартних НА. Кількісне визначення проводять методом абсолютного калібрування з систематичним контролем калібрувального коефіцієнту.
Нижня межа визначення 0,1 мкг/кг.
Методичні вказівки за визначенням N- нітрозамінів затверджені Держкомсанепіднагляду Російської Федерації (МУК 4.4.1.011-93, М., 1993).