

Feinlieb М, Rosenberg НМ, Collins J.G. el al. Trends in COPD morbidity and mortality in the Unated States. Amer. Rev. Respir. Dis., 1989. — 140: 9-18.
Flelcher С, Peto R., Tinker C.M el al. The natural history of chronic
bronchitis and |
emphysema. Oxford. University Press, 1976. — |
119р. |
Flelcher |
С., Peto R The natural history of chronic |
airflow obstruction. |
Br. Med. J., 1977. — 1: 1645-1648. |
|
|
Higgins M.W. Chronic airways disease in the United States: trends and determinants. Chest, 1989. — 96, Suppl. 3:
328-329.
Higgins М. W. Thorn T. Incidence, prevalence, and mortality: intraand inter-country differences. In Clinical Epidemioogy of Chronic Obstructive Pulmonary Disease. Ed. by Hensley M.J., Saunders N.A. — New York, 1990:
23-43.
Jams M.J. Smoking cessation. Eur. Respir. Rev., 1997. — 7 (45): 230-234.
MicNee W, Wedzicha W. Cause of Death in OOPD; still an open question? Monaldi Arch. Chest. Dis., 1997. — 52
(I): 3.
Pauli G.. Kopferichmill MCh.. Spirlet F., Charpin D. Air Pollutants and Allergic Sensitisation. From Genetics to Quality of Life. Proc. of XV World Congress of Astnmology. — Montpellier, Apr. 24-27, 1996. — Ed. Chanez P. et al., 1996.-p. 80-90.
Pope CAIll, Jhun Ш.. Namboodizi MM et al. Paniculate air pollution as a predictor of mortality in a prospective study of US adults. Amer. J. Respir. Crit. Care Med., 1995. — 151; 669-674.
Postma D.Si Epidemiology of COPD: risk factors. In COPD. diagnosis and treatment. Excerpta Medica., 1996. — p. 17.
Report Chronic bronchitis in Great Britain. Brit. Med. J., 1961. — 2: 973.
Safakas KM, Vermeire P.. Pride N.B. et al. Optimal assessment and management of chronic obstructive pulmo-niry disease (COPD) Eur. Respir. J., 1995. — 8: 1398-1420.
Safakas N.M. ERS Consensus Statement: optimal assessment and management of chronic obstructive pulmonary disease. Eur. Respir. Rev., 1996. — 6 (39): 270275.
Thoracic Society of Australia and New Zeiand. Guidelines for management of chronic obstructive pulmonary disease. Mod. Med., August 1995. — 38: 132-146.
Trover G.A, dine M.G., Burrow B. Predictors of mortality in chronic obstructive pumonary disease:a 15 years follow-up study. Am. Rev. Respir. Dis., 1979. — 119: 895-902.
Vermeire P. Definition of COPD. In COPD diagnosis and treatment.Excerpta Medica, 1996. — p. 1-11.
Vermeire P. Guidelines on management of COPD. Eur. Respir. Rev.. — 1997. — 7 (45): 227-229.
Weiss S., Sparrow D. Airway responsiveness and atopy in the development of chronic lung disease. — New York, Raven Press, 1989. —p. 1-19.
Well С Epidemiology of COPD in general practice. In COPD. diagnosis and treatment. Excerpta Medica, 1996.—p. 18-24.
6. Патогенез воспаления при хронических обструктивных болезнях легких
Е. И. Шмелев
Хроническому воспалению принадлежит особая роль в патогенезе хронических обструктивных болезней легких (ХОБЛ). С одной стороны, хроническое воспаление — универсальная реакция на воздействие всех известных (и предполагаемых) факторов риска, с другой — главная
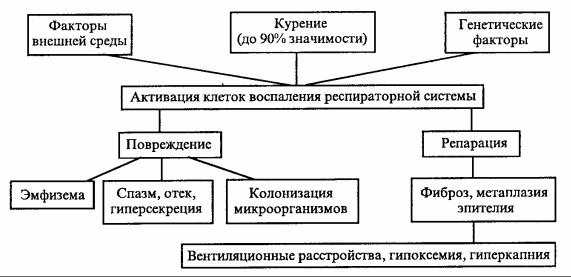
причина всех функциональных и морфологических проявлений ХОБЛ. Преимущественная локализация хронического воспаления, характер его течения определяют индивидуальность ХОБЛ, скорость его прогрессирования, свидетельствуют об адекватности терапии и являются важным прогностическим фактором.
На рис. 6.1 схематично показана последовательность патологических процессов, составляющих хроническое воспаление. Первый этап — воздействие этиологических факторов на клеточные элементы, участвующие в воспалении. Второй этап представлен процессами повреждения и репарации, идущими одновременно и определяющими морфологические и функциональные проявления ХОБЛ. Практически все клеточные элементы респираторной системы под влиянием этиологических факторов активируются и участвуют в воспалительной реакции, которая по сути своей является результатом клеточной кооперации. Но в сложной иерархии межклеточных взаимоотношений, происходящих в разные фазы воспаления, ключевая роль принадлежит нейтрофилам. В норме вся популяция зрелых нейтрофилов условно делится на циркулирующий пул и краевой пул, т. е. клетки, находящиеся в депо. Одним из важных мест депонирования нейтрофилов являются посткапиллярные венулы легких.
Курение ведет к 10-кратному увеличению содержания нейтрофилов в дистальных отделах респираторной системы (MacNee W., 1997). Нейтрофилы, имея средний диаметр 7,03 мкм, постоянно преодолевают микроциркуляторные сегменты альвеол, диаметр которых в среднем равен 5 мкм (Weibel Е. R., 1984), т. е. меньше, чем диаметр нейтрофила. В среднем путь каждого нейтрофила от артериолы к венуле состоит из преодоления 60 капиллярных сегментов. При этом каждая альвеола содержит около 1000 капиллярных сегментов (MacNee W., 1997).
Скорость преодоления капиллярного ложа в значительной мере зависит от способности нейтрофилов к изменению формы, т. е. деформабельности. Под влиянием курения происходит полимеризация актина нейтрофилов, что существенно снижает их деформабельность (Drost Е. et al., 1993). При этом задерживающиеся в капиллярах нейтрофилы окружены очень малым количеством плазмы, несущей антиоксидантный и антипротеолитический потенциал, что создает условия для патогенного действия нейтрофилов.
Место воспаления в патогенезе ХОБЛ
Рис. 6.1
Одновременно с этим механизмом включается следующий, определяемый эндотелием сосудов. Под влиянием воспалительных медиаторов (гистамин, фактор активации тромбоцитов, фактор некроза опухоли, интерлейкин-1 р) усиливается экспрессия адгезивных молекул (Р и Е селектинов) на эндотелиальных клетках, что ведет к адгезии нейтрофилов к сосудистой стенке с помощью специальных рецепторов (Howarth P. H., 1997). После прикрепления к эндотелию нейтрофилы мигрируют через пространства между эндотелиальными клетками, которые увеличиваются под влиянием медиаторов воспаления (Howarth Р. H., 1997). Направленность движения нейтрофилов определяется различными хемоаттрактантами, самым сильным из которых является интерлейкин-8 — ИЛ-8 (Stockley R. А., 1997). Помимо ИЛ-8 хемоаттрактантами для

нейтрофилов служат LT-B4, фактор активации тромбоцитов, С5 и никотин (Repine J. Е. et al., 1997).
Нейтрофилы, проникая в межклеточное пространство, в полной мере проявляют свой патогенный потенциал. Наряду с вьщелением ряда противовоспалительных медиаторов, обладающих хемотаксическим действием для других нейтрофилов (ФАТ, LT-B4,12-НЕТЕ, вазоактивные простагландины Ег и Р^а), нейтрофилы выделяют ряд субстанций, обладающих мощным деструктивным потенциалом, распространяющимся практически на все молекулярные компоненты тканей: липиды, белки, нуклеиновые кислоты. Это, в первую очередь, нейтральные протеазы (эластин) и кислородные радикалы (Rennard S., 1997; Stockley R. A., 1997). Помимо этого, катионные белки и сс-глюкорунидаза нейтрофилов обладают цитопатогенным действием. В норме весь этот комплекс патогенных компонентов направлен на разрушение чужеродных субстанций, попавших во внутреннюю среду. Но под влиянием факторов риска ХОБЛ, главным из которых является курение, запускается механизм (описанный выше), извращающий защитное действие нейтрофила.
В условиях гигантского скопления нейтрофилов в капиллярной сети альвеол, где местный антипротеазный потенциал, определяемый плазменными фактора-' ми, быстро истощается, происходит разрушение структурных элементов альвеол и формирование эмфиземы. В первую очередь разрушаются участки альвеолярных стенок, прикрепляющиеся к терминальным бронхиолам. Помимо этого, оксиданты и другие компоненты табачного дыма могут инактивировать α1- антипротеазный ингибитор (Gadek J. Е. et al., 1979), тем самым усугубляя локальный дефицит антипротеаз. Помимо этого, эластаза разрушает эпителиальные клетки, ведет к метаплазии бокаловидных клеток (Rennard S., 1997).
Второй очень важный элемент патогенного воздействия нейтрофилов — оксидативный стресс, т. е. выделение непомерно большого (превышающего физиологические потребности) количества свободных радикалов, обладающих мощным повреждающим действием (рис. 6.2).
Роль оксидативного стресса в патогенезе ХОБЛ
Рис. 6.2
Кислород, озон, NО2 оказывают мощное оксидантное действие на все структуры респираторной системы (Repine J. Е., et al., 1997; Stockley R. A., 1997). Сигаретный дым — наиболее опасный и изученный инициатор оксидативного стресса в легких. Основными оксидантами табачного дыма являются О2, О3, ОН, Н2О2, NО, НОСl2. Реализацию своего действия оксиданты осуществляют при участии ионов железа в качестве катализатора. Известно, что при ХОБЛ в легких, и особенно в альвеолярных макрофагах, скапливается большое количество железа (Tompson А. В. et al., 1991).
Наиболее изученным экзогенным источников оксидантов является курение, а эндогенным — нейтрофилы и альвеолярные макрофаги (Repine J Е et al 1997-Stockley R. A., 1997).
Легочная антиоксидантная защита состоит из энзимных и неэнзимных систем (James D., 1993). Главные энзимные антиоксиданты — супероксиддисмутаза (СОД) и глутатион (Г). Витамин Е, β-каротин, витамин С, мочевая кислота, флавоноиды, билирубин — представители неэнзимной антиоксидантной системы (11).
Распределение антиоксидантов в плазме крови и на поверхности эпителия бронхов представлено в табл. 6.1 (Bast A., 1996).
Таблица 6.1
Антиоксиданты |
Плазма, мкл |
Бронхиальное |
|
содержимое, мкл |
|||
|
|
||
|
|
|
|
Витамин С |
40 |
100 |
|
|
|
|
|
Глютатион |
1,5 |
100 |
|
|
|
|
|
Мочевая кислота |
300 |
90 |
|
|
|
|
|
Билирубин |
10 |
— |
|
|
|
|
|
Токоферол |
25 |
2,5 |
|
|
|
|
|
-каротин |
0,4 |
— |
|
|
|
|
|
Альбумин-SH |
500 |
70 |
|
|
|
|
Как следует из приведенных данных, глютатион и витамин С являются главными внутриклеточными антиоксидантами, и их эндобронхиальное содержание значительно превосходит концентрацию в плазме.
Оксиданты оказывают прямое токсическое действие на ключевые структуры легких: соединительную ткань, ДНК, липиды, протеины (Repine J. Е., 1997). Оксиданты усиливают синтез гликоконъюгатов слизи эпителиальными клетками (Adier К. В. et al., 1990), нарушают функции ресничек (Feldman С. et al., 1994). Оксиданты стимулируют формирование тромбоксанов, уменьшают активность сурфактанта, повреждают фибробласты, усиливают эндотелиальную проницаемость (Repine J. Е., 1997).
Оксиданты инактивируют ингибиторы протеаз (Carp M., 1982). В ответ на это эластаза разрушает альвеолярные стенки, экстрацеллюлярные мембранные протеины, стимулирует синтез ИЛ-8.
Таким образом, оксидативный стресс оказывает разностороннее повреждающее действие практически на все легочные структуры, и выраженность его регулируется состоятельностью антиоксидантных систем.
В последнее десятилетие интенсивно исследуется роль NО в патогенезе болезней легких. NО синтезируется из L-аргинина под влиянием NO-синтаз (NOS) при участии кальция и кальмодулина (Sing S., Ewans Т. W., 1997). Известны 3 формы NOS: эндотелиальная, макрофагальная и нейтронная (Sing S., Ewans Т. W., 1997), которые ведут к локальному синтезу NО и определяют его влияние на респираторную систему.
NО, выделяемый эндотелиальными клетками, обладает сосудорасширяющим действием на уровне мелких артерий и артериол, регулируя сосудистое сопротивление (Radomski M. W. et al., 1987; Griffiths M. J. D. et al., 1994). При этом установлено, что гипоксия снижает синтез NO (Vallance P., Collier J., 1994).
NО ингибирует адгезию (Radomski M.W., 1987), активацию и агрегацию тромбоцитов, препятствуя внутрисосудистому тромбообразованию (Azuma H. et al., 1986). NO обладает прямым бронходилатирующим действием (Lammers J. W. J. et al., 1992), а также нейтрализует бронхоконстрикторное влияние ацетилхолина. Макрофагальный NO участвует в противоинфекционной защите путем усиления внутриклеточного разрушения микобактерий туберкулеза и других микроорганизмов. NO усиливает функцию реснитчатого аппарата и способствует стерилизации респираторного тракта. Способность альвеолярных макрофагов продуцировать NO играет важную роль в поддержании местного иммунного гомеостаза респираторного тракта (Holt P. G., 1995).
Процессы повреждения и репарации, происходящие при ХОБЛ, составляют суть хронически текущего воспаления, регулируются громадным количеством экзогенных и эндогенных противовоспалительных медиаторов и проявляются на всех уровнях респираторной системы: уровень защитной слизи, уровень эпителиальных клеток и интерстициальная ткань.
Слизь, покрывающая эпителий, является постоянно действующим фильтром, разделяющим воздух и поверхность эпителиального слоя. Этот барьер транспортабелен, постоянно обновляется, способен нейтрализовать патогенное действие токсических и инфекционных факторов. Мозаичный набор гликопротеиновых рецепторов муцина слизи в норме распознает и фиксирует микроорганизмы, которые затем элиминируются мукоцилиарно-транспортным механизмом (Girod S. et al., 1992).
Под влиянием этиологических факторов нарушаются реологические свойства слизи, соотношения фракций гель/золь, снижается противоинфекционный потенциал. Это все нарушает клиренс и лишает эпителий первой линии защиты (PuchelleE.etal.,1997).
Повреждение и регенерация альвеолярного эпителия при ХОБЛ — постоянно идущий многокомпонентный процесс, который в общих чертах характеризуется следующими закономерностями:
1)Миграция базальных клеток в зоны повреждения.
2)Восстановление плотных межклеточных соединений.
3)Дифференциация клеток в направлении сквамозной и бокаловидной метаплазии (Trevisanil et al., 1990; Jeffery P. K-, 1992). Метаплазия бокаловидных клеток в первую очередь проявляется увеличением их числа и гиперсекрецией (Spurzem J. R. et al., 1991). Сквамозная метаплазия происходит из клеток, потерявших контакт с базальной мембраной и напоминающих либо клетки пищевода, либо кожи.
4)Активные митозы, ведущие к гиперплазии базальных и мукозных клеток (Puchelle Е. etal., 1997).
Естественно, что регенерация эпителия происходит с участием белков экстрацеллюлярного матрикса. Фибронектин (Spurzem J. R. et al., 1991) создает первичное ложе для клеток; 1 У тип коллагена и ламинин действуют как якорные молекулы для мигрирующих клеток. После повреждения клетки должны быть удалены.
Большую роль в удалении этих клеток играют матриксные металлопротеазы — семейство энзимов, разрушающих практически все компоненты экстрацеллюлярного матрикса — желатиназы и стромелизины (Murphy G., Etocherty A. J. P., 1992). Особое место среди эпителиальных клеток занимают клетки Клара (Singh G, Katyal S., 1997), которые являются важным источником репаративных процессов (KhoorA. et al., 1997), а секреторные протеины этих клеток обладают противовоспалительной активностью.
Повреждение эпителия создает благоприятные условия для имплантации микроорганизмов. У обнаженного матрикса есть рецепторы к бактериям, что создает условия для колонизации микрофлоры (Chanez P. et al., 1997).
В экстрацеллюлярном матриксе происходят аналогичные процессы. После оксидативного или протеолитического повреждения интерстиция происходит пролиферация фибробластов. В поздних стадиях хронического воспаления преобладает фиброз с отложением экстрацеллюлярного матрикса в стенках бронхиол (Pare P. D. et al., 1997). Есть даже предположения о том, что перибронхиальный фиброз играет большую роль в обструкции, чем эмфизема (Puchelle Е. et al., 1997).
На разных стадиях дифференциации клеток в процессе репарации, а также и во внеклеточных субстанциях усиливаются рецепторы к цитокинам, являющимися основными регуляторами репарации. Цитокины продуцируются в основном альвеолярными макрофагами, нейтрофилами, эпителиальными клетками, лимфоцитами и фибробластами.
Трансформирующий фактор роста ((TgF-β), фактор роста тромбоцитарного происхождения (PDgF), гранулоцитарно-макрофагальный колониестимулирующий фактор (gmCSF) и основной фактор роста фибробластов (L FgF) — основные фиброгенные цитокины (Chanez P. et al., 1997). При этом в межклеточных взаимоотношениях важнейшими факторами являются время и концентрация цитокина. При изменении этих условий один и тот же цитокин может оказывать диаметрально противоположное действие (Dros Е. et al., 1993; Chanez P. et al., 1997).
В результате хронического воспаления происходит ремоделирование бронхов, которое проявляется:
1)Увеличением подслизистого и адвентициального слоя (отек, отложение протеогликанов, коллагена).
2)Увеличение размеров и числа слизистых и бокаловидных клеток.
3)Увеличение бронхиальной микрососудистой сети.
4) Гипертрофия и гиперплазия мускулатуры воздухоносных путей (Pare P. D. et al.,
1997).
Инфекционный процесс занимает особое место в патогенезе воспаления при ХОБЛ. Действие инфекционных агентов (бактерии, вирусы, грибы) многогранно, и условно можно выделить следующие компоненты: повреждающее, провоспалительное и иммуномодулирующее.
Отношение врачей к значимости инфекций в генезе рецидивов ХОБЛ базируется обычно на результатах бактериологического исследования мокроты и дефекта антибактериальной терапии. Однако к результатам бактериологического исследования мокроты обычно примешивается бактериальный пейзаж ротоглотки из-за загрязнения бронхиального содержимого при получении материала (RiiseG.S., 1997).
При исследовании большой популяции больных ХОБЛ установлено, что антибактериальная терапия не уменьшает числа рецидивов (Riise G. S., 1997), а обострение ХОБЛ не является синонимом внутрилегочной инфекции (Murphy Т. J., Sethi S., 1992).
Около 1/3 больных ХОБЛ подвергаются рецидивированию респираторной инфекции с нарастанием бронхиальной обструкции. При этом наиболее частыми бактериальными агентами являются Strept. pneumonia, Haemophilus influenzae и Moraxella catarralis (Siafakes N. M. et al., 1995; Riise G. S., 1997).
Наряду с бактериальными агентами около 1/3 причин инфекционных рецидивов составляют вирусы, чуть меньше — микоплазмы и хламидии(Магипег J., 1997; Riise G. S., 1997). В то же время при стабильном течении ХОБЛ (т. е. вне обострения) специальными бронхологическими методами у 50% больных установлена колонизация слизистой бронхов нормальной орофарингеальной флорой (Irwin R. S. et al., 1982; Martinez J., 1997). Следовательно, следует выделять колонизацию микроорганизмов и инфекцию (Murphy Т. J., Sethi S., 1992). Этот краткий перечень фактов свидетельствует о существовании определенных разночтений в понимании роли инфекции в патогенезе ХОБЛ. Итак, повреждающее действие инфекционных агентов. В эксперименте установлено, что острая вирусная инфекция ведет в трехдневный срок к полной десквамации эпителия (Puchelle Е. et al., 1997). Регенерация начинается с пятого дня поражения. В этот период создаются благоприятные условия для имплантации микроорганизмов. Тем более, что у обнаженного матрикса есть рецепторы к микроорганизмам. Так, ламинин и фибронектин (Puchelle Е. et al., 1997) связывают Pseudomonas aeruginosa. Клетки, мигрирующие в зону повреждения, также имеют рецепторы к микроорганизмам (Puchelle Е. et al., 1997). Роль курения, как и других факторов риска ХОБЛ, в колонизации микроорганизмов показана в ряде целенаправленных исследований. Так, R. S. Irwin et al.(1988) при 2-летнем мониторировании курящих лиц с ХОБЛ с использованием специальной технологии, исключающей контаминацию орофарингеальной флорой, установили, что курение более 1 пачки сигарет в день коррелирует с эндобронхиальной колонизацией микроорганизмов. У больных, прекративших курить, колонизация уменьшается, клиренс быстро восстанавливается.
Е. Monso et al. (1995) показали, что у четверых больных ХОБЛ вне обострения обнаруживается колонизация микроорганизмов в нижних дыхательных путях. Причем наиболее часты находки Н. influenzae и S. pneumoniae. В то же время при обострениях, связанных с инфекцией, у этих больных частота положительных культуральных результатов удваивается (~ 50%), но не достигает 100%. Это подчеркивает определенные расхождения в клинической картине и результатах бактериологического исследования.
Колонизирующиеся микроорганизмы проявляют целый ряд вирусных факторов, вносящих свой вклад в воспаление: 1) стимуляция продукции слизи; 2) цитотоксичность; 3) инвазия в эпителий за счет адгезий; 4) выделение IgA протеиназ; 5) продукция гистамина (Van Alpphen L., 1995).
Естественно, что колонизация микроорганизмов является мощным аттрактантным стимулом для циркулирующих и оседлых фагоцитов, что ведет к усилению оксидативного стресса и наращивает протеолитическую деструкцию не только микроорганизмов, но и зон их колонизации. Одним из важных условий колонизации является изменчивость антигенных структур микроорганизмов, т. н. мимикрия, что создает трудности в распознавании микроба и его эффективного удаления (Van Alpphen L., 1995).
На основании анализа взаимоотношений между микроорганизмами в респираторном тракте и защитными механизмами, Р. Cole и R. Wilson в 1989 г. опубликовали свою гипотезу порочного круга, смысл которой заключается в том, что персистенция микроорганизмов ведет к повреждению факторов защиты, а нарушение факторов защиты способствует колонизации микробов. Существенная роль в инициации этих хронических процессов придается вирусному повреждению. Колонизация стимулирует воспаление, которое приобретает хроническое течение.
J. Hogg (1997) приводит данные вирусологического обследования больных ХОБЛ. У 262 было сочетание положительных культуральных исследований и нарастание титра антител.
Статистически достоверная связь позитивных лабораторных тестов с нарастанием респираторной симптоматики отмечена при инфекции риновирусом, вирусом гриппа, парагриппа. Риносинцитиальный вирус, аденовирус, простой герпес при положительных результатах культурального и серологического тестирования не влияли на выраженность клинических симптомов. В итоге из 262 больных с лабораторными признаками вирусной инфекции лишь у 181 она ассоциировалась с обострением ХОБЛ, а у 81 не установлено непосредственного участия в обострении болезни, т. е. так называемая латентная инфекция. Автор, исследуя ДНК аденовирусов с помощью полимерной цепной реакции, предполагает, что латентная вирусная инфекция усиливает провоспалительное действие курения. Основную роль в этом играет EIA протеин, продуцируемый ДНК персистирующего вируса. Экспрессия EIA гена на клетках респираторного эпителия усиливает продукцию провоспалительных цитокинов этими клетками, в частности, внутриклеточные адгезивные молекулы-1 и ИЛ-8.
Итак, отчетливо формируются 2 формы сосуществования микроорганизма и хозяина при ХОБЛ: колонизация и инфекция. Разграничить в каждом конкретном случае эти два состояния бывает трудно, но для определения стратегии лечения и выбора антибактериальных средств это очень важно.
Специальные исследования взаимоотношения процессов колонизации и инфекции проливают свет на решение этого сложного вопроса. Известно, что наличие хронической трахеостомы является важным фактором риска инфекций респираторного тракта. R. Huriid et al. в 1996 г. при наблюдении в течение года за 39 больными, имеющих трахеостомы, в домашних условиях у 38 обнаружена колонизация трахеи патогенами, но у большинства (70%) бронхиальное содержимое было стерильным. И в течение года лишь у 18 пациентов возникали эпизоды респираторной инфекции нижних дыхательных путей. Это наблюдение свидетельствует об определенной дистанции между колонизацией и инфекцией.
Однако несомненно, что колонизация является важным условием для возникновения инфекции. Сама по себе колонизация микроорганизмов в нижних отделах респираторного тракта является индикатором местного дефекта защиты бронхов и вносит существенный вклад в хроническое течение воспаления (Cole P., Wilson R, 1989; Van Alphen L., 1995).
Таким образом, хроническое воспаление при ХОБЛ является ключевым элементом прогрессирования заболевания. Воспаление захватывает не только все слои бронхиальной стенки, но и интерстициальную ткань. Воспаление ведет к формированию основных морфологических проявлений ХОБЛ: эмфизема легких, ремоделирование воздухоносных путей, включая и перибронхиальный фиброз.
Основными компонентами патогенеза хронического воспаления являются оксидативный стресс, протеолитическая деструкция ткани, проявляющаяся под влиянием факторов риска ХОБЛ. Основными клетками-эффекторами являются нейтрофилы, действие которых усиливается другими элементами респираторной системы, а также персистирующими микроорганизмами.
ЛИТЕРАТУРА
Adier К.В., el al. Oxygen metabolites stimulate release of high molecular weight glycoconjugates by cell and organ cultures of rodent respiratory epithelium via an orachidonic acid-dependent mechanism. S. din. Invest. 1990. 85: 7585.
AzwnaH, et al. Endothelium-dependent inhibition of platelet aggregation. Br. J. Pharmacol. 1986. 88: 411-415.
Bast A. Oxidants and antioxidants in the lung. Exept. Med. 1996, 33-39.
Carp M, et al. Potential mechanism of emphysema. Proc. Nau. Acad. Sci. — USA, 1982. — 79: 2041-2045.
Chanez P., et al. Remodelling of the airways in chronic obstructive pulmonary disease. Eur. Resp. Rev. 1997. — 7,43: 142-145.
Cole P., mison R. Host-microbial interrelationships in respiratory infection. Chest. 1989. — 217S-221S.
Cosio M, et al. The relations between structural changes in small airways and pulmonary function tests Curr Microbiol. 1993. — 26: 91-95.
Drost E., et al. Deacreased leukocyte defomiaKlity following acute cigarette smoldng in smokers. Am. Rev. Respir. Dis. 1993.— 148: 1277-1283.
Fetdman С, et at. Oxidant-mediated ciliary dysfunction. Free Radio. Bid Med. 1994. — 17: 1-10. Gadeic J.E.. Fells G.A, Crystal RG. Cigarette smoking in-
duces antiprotease deficiency in the lower respiratory tract of humans. Science. 1979.—206: 1315-1316.
Girod S., et al. Role of the physico-chemical properties of mucus in the protection of the respiratory epithelium. 1992.—S 477-487.
Griffiths M.J.D., et al. Nitric oxide synthase inhibitors in septic shock, din. Intens. Care. 1994. — 5: 29-36.
Harlid R., el al. Respiratory tract colonization and infection in patients with chronic tracheostomy. Am. J. Respir Crit. Care Med. 1996. — 154: 124-129.
Heffsr J.A., Repine J.E. State of art: pulmonary strategies antioxidant defense. Am. Rev. Respir Dis 1989 — 140:531-554.
Hogg J. Latent adenoviral infections in the pathogenesis ofOOPD. Eur. Respir. Rev. 1997. — 7, 45: 216-220.
Holt P.G. Inflammatory responses in airway tissues: which cell types have been implicated, in book. Proceed Symp. — London. Nov. 10, 1995. 8-14.
Howarlh P.H. What is the nature of asthma and where are the therapeutic targets? Respir. Med. 1997. — Suppl. A, 2-8.
Irwin RS., et al. Prediction oftracheobronchial colonization in current cigarette smokers with chronic obstructive bronchitis. J. Infect. QS. 1982. — 145: 234-241.
James D„ et al. The control of neutrophil chemotaxis by inhibitors of cathepsin G and chymotrypsin. J. Bid Chem. 1993. — 270: 23437-23443.
Jeffery P.K. Hstological features of the airways in asthma and COPD. Respiration. 1992.—1: 13-16. Khoor A., et al. Ontogeny of Clara cell-specific protein and its mRNA: their association with neuroepithelial bodies in human fetal lung and in bronchopulmonary displasia. J. Hstochem. Cytochem. 1996. — 44: 14291438. Larnmers J. W.J., et al. Nonadrenergic, nonchdinergic airway inhibitory nerves. Eur. Resp. J. 1992. — S, 239-246. MacNee W. Neutrophil traffic and COPD. Europ. Resp. Rev. 1997. — 7, 43: 124-127. Martinez J. Antibiotics and vaccination therapy in COPD. Eur. Resp. Rev. 1997. — 7, 45: 240-242.
Monso E., et al. Bacterial infection in chronic obstructive pulmonary disease Am J Resp Crit Care Med 1995.—152: 1316-1320.
Murphy G., DochenyAJ.P. The matrix metalloproteinases and their inhibitors. Am. J. Respir. Cell Molec. Biol. 1992.—7: 120-125.
Murphy T.J., Sethi S. Bacterial infection in chronic obstructive pulmonary disease. Am. Rev. Respir. Hs. 1992. — 146: 1067-1083.
Pare P.O., et a\. Pathophysiological process in chronic obstructive pulmonary disease, in book The Role of An-ticholinergics in COPD and Chronic Asthma.
— London, 1997. 19-30.
Plolkonstd M.C.. et al. High affinity of Pseudomonas aeruginosa with laminin. Infect, hnmun. 1996. — 64: 600-605.
Plotkowsld M.C, et al. Pseudomonas aeruginosa binds to sollude cellular fibronectin. Curr. Microbiol. 1993. — 26:91-95.
Puchelle £, et al. Airway epithelium injury and repair. Eur. Resp. Rev. 1997. — 7,43: 136-141.
Radomski M. W., et al. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 1987.ii 1057-1058.
Bernard S. Pathophysiological mechanisms of COPD. Eur. Resp. Rev. 1997. — 91, Suppl. A: 2-8.
Repine J.E., et al. Oxidative stress in chronic obstructive Pulmonary Disease. Am. J. Respi. Crit. Care Med. 1997.—V.I 56: 341-357.
Riise G.S. Bacterial cdonization in chronic bronchitis and COPD. P. News. 1997. — 1: 13-15.
Siafakes N.M., el al. Optimal assessment and management of chronic obstructive pulmonary disease (COPD). Eur. Resp.J. 1995.—8: 1398-1420.
Sng S, Evans T.W. Nitric oxide, the bidogical mediator of decade: fact or fiction? E. Resp. J. 1997. — 10:
699-704.
Sngh G.. Kalyal S.L Clara Cells and Clara cell lOkD protein. Am. J. Respir. Cell Ш. Bid. 1997. — v.l7:
141-143.