

4.Формирование зачатков осевых органов (головного и спинного мозга, позвоночного столба, первичной кишки) и формирование плаценты (3-8-я неделя развития)
После завершения имплантации в развитии эмбриона начинается четвертый критический период – период образования зачатков органов и систем эмбриона, который у человека заканчивается к третьему месяцу внутриутробной жизни. Этот срок является наиболее чувствительным для развития эмбриона (3-7 недель), а также период образования плаценты (9-12 недель). В период образования органов, в результате патогенного действия повреждающих факторов на эмбрион, в первую очередь, поражаются те органы и системы, которые находились в это время в процессе закладки и повышенного обмена веществ. Поэтому для поражения эмбриона в этот период характерным является возникновение уродств (тератогенный эффект). У эмбриона человека наиболее чувствительными к неблагоприятным факторам являются центральная нервная система, органы зрения, железы внутренней секреции и половые железы, поэтому аномалии этих органов встречаются чаще других.
Под воздействием повреждающих факторов во время образования плаценты происходят патологические изменения, в основе которых лежит возникновение плацентарной недостаточности, выражением которой является врожденная гипотрофия плода (дефицит массы тела ребенка).
После завершения процессов формирования органов и формирования плаценты начинается плодовый период (с конца второго - начала третьего месяца беременности до рождения). Эмбриотоксического и тератогенного * эффектов, столь выраженных и типичных для предыдущий стадий, в этот период не наблюдается. Исключение составляют лишь аномалии развития половых органов у плодов женского пола, так как половые органы начинают формироваться в относительно поздний период (12 –14 неделя эмбрионального развития). Это может произойти при приеме гормональных препаратов, в том числе и оральных контрацептивов:
Эстрогены и эстроген + гестогеносодержащие препараты - наиболее уязвимые для срока беременности от 8 до 17 недель. Результатом их применения может быть развитие опухолей гениталий, анатомических и функциональных их дефектов, а также возможно нарушение полового развития у плода мужского пола, - псевдогермофродитизм.
Прогестероносодержащие пероральные контрацептивы - их воздействие возрастает от начала беременности до 4-х месяцев. Результатом их применения может быть нарушение формирования половых органов у плода и псевдогермафродитизма девочек, а также возможны аномалии конечностей, сердечно-сосудистой системы, желудочно-кишечного тракта.
Аномалии развития в этот период именуются эмбриопатиями.
Эмбриопатия - патология эмбрионального периода с 16-го дня беременности до 75-го дня включительно, в течение которого заканчивается основной органогенез и формирование амниона и хориона. К основным видам эмбрионатий относят врожденные пороки развития. Врожденным пороком развития называют стойкое морфологическое изменение органа, части тела или всего организма, выходящее за пределы вариаций нормального строения определенного биологического вида, возникащее внутриутробно в результате нарушений морфогенеза. Так как органогенез завершается в основном в эмбриональный период, большинство пороков развития появляется именно на этом этапе внутриутробного существования. Однако, кроме врожденных пороков с нарушениями основного морфогенеза органов или частей тела, имеются врожденные пороки, при которых нарушения развития наблюдаются на уровне тканевой дифференцировки. Они часто бывают системными, например, пороки развития поперечно-полосатой мускулатуры (врожденная миатония Оппенгейма), соединительной ткани (болезнь Марфана), кожи (врожденный ихтиоз), костей хрящевого генеза (врожденная хондродисплазия) и др. Пороки развития могут касаться также тканей одного органа, например гипоплазия гладкой мышечной ткани при megaureter, нервного интрамурального аппарата - при mеgасоlоп, легочной ткани - при кистозном легком и др. По срокам возникновения эти пороки относятся к ранним фетопатиям. Ранние фетопатии часто сочетаются с эмбриопатиями; например, врожденный ихтиоз и хондродисплазия - с порокам развития лица, болезнь Марфана с пороками развития лица и аорты и др. Частота врожденных пороков, по данным ВОЗ, составляет 1,3% от общего числа рождений. Любой врожденный порок может проявляться в виде:
1) отсутствия какоголибо органа или части тела (агенезия, аплазия);
2) недоразвития органа (гипоплазия);
3) чрезмерного развития (гиперплазия) или наличия избыточного числа органов (удвоение и др.);
4) изменения формы (слияние органов, атрезия, стеноз отверстий, каналов, дизрафия - незаращение эмбриональных щелей, экстрофия - выворот и др.);
5) изменения в расположении органов (эктопия);
6) персистирования эмбриональных провизорных (предсуществовавших) органов.
Классификация. Врожденные пороки развития разделяют по степени распространенности в организме, по локализации в том или ином органе, по этиологии. Пораспространенности врожденные пороки могут быть:
1) изолированными - с поражением одного органа;
2) системными - с поражением нескольких органов одной из систем;
3) множественными - с поражением органов разных систем.
По локализации различают пороки развития центральной нервной, сердечно-сосудистой, пищеварительной, мочеполовой и других систем. Врожденные пороки развития названной локализации имеют наибольшее значение в патологии. Чаще всего встречаются пороки развития центральной нервной и сердечно-сосудистой систем, так как именно эти системы имеют наибольший тератогенный терминационный период. Изолированные пороки развития встречаются чаще множественных, несмотря на то что тератогенный терминационный период для многих органов во времени совпадает. Наиболее совершенной является классификация пороков развития по этиологии, однако уровень современных знаний пока не позволяет ее придерживаться. Однако известны отдельные виды системных и множественных врожденных пороков, связанных с определенной этиологией, например рубеолярная эмбриопатия, алкогольная, талидомидная эмбриопатии и др., а также наследственно обусловленные генотипические врожденные пороки и врожденные пороки вследствие хромосомных аберраций; последние, как правило, носят характер множественных. Разграничение генотипических врожденных пороков с их фенокопиями возможно с помощью генеалогического метода изучения родословной, цитогенетического метода, позволяющего изучить кариотип тканей носителя порока при их культивировании, с помощью близнецового метода, основанного на частоте выявления врожденных пороков у однояйцевых близнецов и метода дерматоглифики - изучения комплекса кожных узоров, расположенных на ладонях, подошвах и сгибательной поверхности пальцев, который используется для срочной диагностики хромосомных болезней.
Миатомия Оппенгейма-врожденная амиотония, относится к группе врожденных поражений нервно-мышечной системы. В основе заболевания, по мнению большинства авторов, лежит дефект развития клеток передних рогов спинного мозга. В настоящее время многие не считают миатонию Оппенгейма самостоятельной нозологической формой, а рассматривают ее как доброкачественный вариант ранней формы болезни Верднига-Гоффманна. Не исключены также дефекты внутриутробного развития спинного мозга и мышечной ткани.
При гистологическом исследовании находят изменения в передних рогах, особенно в поясничном утолщении. Наблюдается резкое уменьшение количества ганглиозных клеток. Имеющиеся клетки малы, бедны хроматином, с резко уменьшенной протоплазмой, с отсутствием аксонов или резким изменением их формы. Передние корешки истончены. Аналогичные изменения найдены в больших пирамидных клетках Беца и некоторых ядрах черепных нервов. В мышечной ткани отмечаются значительные изменения, большая часть волокон подвергается дегенерации. Тонкие пучки мышечных волокон разделяются небольшими скоплениями жира и фиброзной соединительной тканью.
Симптомы заболевания выявляются уже в первые дни жизни. В некоторых случаях дефект двигательной сферы отмечается еще внутриутробно (слабое шевеление плода). Все движения ребенка совершаются вяло, в тяжелых случаях конечности неподвижны, на ощупь мышцы мягкие за счет резкого снижения мышечного тонуса, который и является ведущим симптомом и обусловливает в основном двигательный дефект. Иногда гипотония достигает степени полной атонии. Конечностям можно придавать любое положение. Отмечается расслабленность связочного аппарата. Сухожильные рефлексы угнетены. Двигательные расстройства обычно симметричны, более резко выражены в нижних конечностях. Чувствительность сохранена. Психика не изменена. Дети, как правило, хорошо упитаны. Электромиография выявляет изменения, характерные для поражения передних рогов.
Течение заболевания стационарное или отмечается некоторое улучшение состояния, увеличение объема движений, больные могут держать голову, сидеть, иногда стоять и даже передвигаться. Дети редко доживают до 14-15 лет, смерть наступает от интеркуррентных заболеваний (чаще от поражения легких).
Дифференциальный диагноз проводят с острым полиомиелитом, в отличие от которого при миатонии наблюдаются симметричные поражения мышц, слабо выражены мышечные атрофии. Рахит сопровождается выраженной мышечной гипотонией, но в этом случае наблюдаются и другие признаки рахита.
Лечение заболевания. Назначают АТФ, глютаминовую кислоту, витамины B1, E, а также галантамин, препараты анаболических гормонов,. Показаны систематические курсы массажа, ЛФ>К- Прогноз малоблагоприятный.
Синдром Марфана (Болезнь Марфана, Marfan syndrome) — аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван мутациями генов, кодирующих синтез гликопротеина фибриллина-1, и является плейотропным. Заболевание характеризуется различной пенетрантностью и экспрессивностью. В классических случаях лица с синдромом Марфана высоки (долихостеномелия), имеют удлиненные конечности, вытянутые пальцы (арахнодактилия) и недоразвитие жировой клетчатки. Помимо характерных изменений в органах опорно-двигательного аппарата (удлинённые трубчатые кости скелета, гипермобильность суставов), наблюдается патология в органах зрения и сердечно-сосудистой системы, что в классических вариантах составляет триаду Марфана.
Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40 годами, и смерть наступает вследствие расслаивающейся аневризмы аорты или застойной сердечной недостаточности. В странах с развитым здравоохранением больные успешно лечатся и доживают до преклонного возраста.
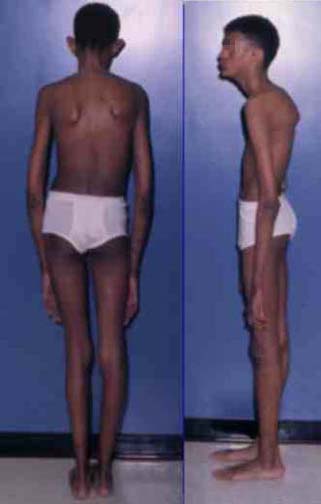
Рис 6.Внешний вид больного с синдромом Марфана.