

Лабораторная работа 3
1. Построение кривых потенциальной энергии молекулы
Задание 1.
1. Построить молекулу этана. В пункте меню Setup укажите, что для вычислений будет использоваться метод молекулярной механики MM+.
2. Рассчитайте энергию молекулы (Соmpute, Single Point). После выполнения расчета энергия молекулы будет выведена в строке статуса (2,36 ккал/моль).
3. Минимизируйте энергию молекулы (Соmpute, Geometry Optimization). Отметьте, что энергия молекулы при этом снизилась до 0,82 ккал/моль).
4. Создайте log-файл, для записи вычислений с помощью команд File, Stat Log. В открывшемся окне укажите директорию, где будет находиться файл и имя файла. Не записывать в директорию С/Sample!!!
5. Пронумеруйте атомы в молекуле и выберите торсионный угол. Выполните команду Соmpute, Роtential. Сделайте следующие установки в окне:
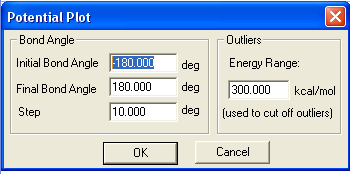
Это означает, что выделенный вами торсионный угол будет меняться от 180 до -180 с шагом 10.
6. После выполнения вычислений на экран будет выведена кривая потенциальной энергии.
7. Закройте log-файл File, Stop Log. Перенесите данные файла в Excel. Постройте кривую потенциальной энергии этана в Excel.
Задание 2.
1. Сделать расчет кривой потенциальной энергии для бутана.
Задание 3. Рассчитать кривую потенциальной энергии для нуклеотида (молекула выдается преподавателем).
1. Для построения молекулы использовать команды Databases, Nucleic Acids.
Важнейшей чертой пространственной структуры нуклеозидов и нуклеотидов является взаимное расположение остатков основания и углевода, которое обычно описывается двумя параметрами.
Первый параметр– величина двухгранного угла между плоскостями оснований и углеводного остатка (в этом случае рассматривается средняя квадратичная плоскость фуранозного кольца). Для всех нукдеозидов и нуклеотидов этот угол приближается к прямому, хотя никогда и не достигает его (от 55 для аденозин–5–фосфата до 81 для 5–иоддезоксиуридина).
Второй параметр– величина угла поворота основания вокруг гликозидной связи .
|
Принятая нумерация для гетероциклических оснований |
|
|
|
|
тимин (5-метил, 2,4 диоксипиримидин) |
аденин (6–аминопурин) |
|
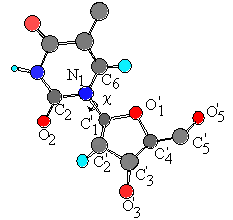
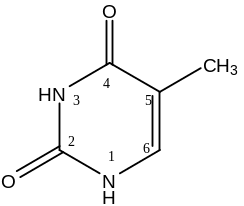
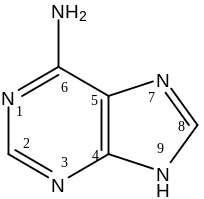
Угол принято характеризовать как угол вращения относительно связей С1N1 в пиримидиновых или С1N9 в пуриновых остатках. Угол принимаеися равным 0, когда связи С1O1 и N1С6 (в пиримидиновых производных) или С1O1 и N9С8 (в пуриновых производных) находятся в цис-положении.
Детальный анализ конформаций пиримидиновых и пуриновых нуклеозидов и нуклеотидов позволил выделить две области с наименьшими внутримолекулярными взаимодействиями.
Первая область лежит при значениях в районе –30, вторая – в районе +150. Конформации, отвечающие первой области, принято называть анти-конформациями, соответствующие второй области– син-конформациями.
Для пиримидиновых нуклеозидов в случае анти-конформации ближе всего с атому расположен атом водорода при С6 , а в случае син-конформации – атом кислорода при С2.
|
|
Антиуридин |
Синуридин |
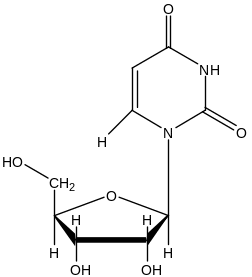
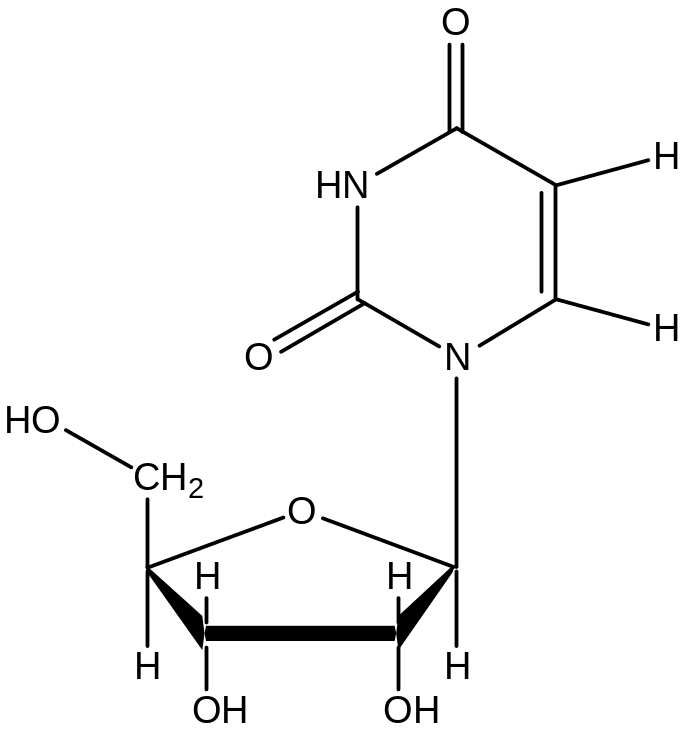
У
антиуридин
|
|
Антиаденозин |
Синаденозин |
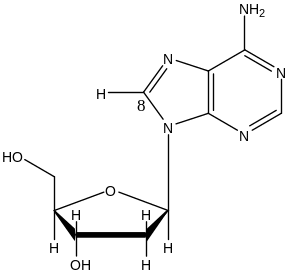
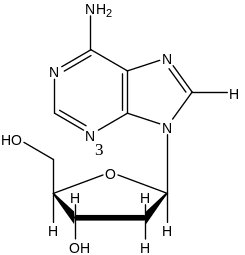
2. Установить, какие конформации (анти- или син-) наиболее характерны для вашей молекулы. Построить молекулы с торсионными углами, соответствующим минимумам энергии.
Задание 4. Построение кривой потенциальной энергии для циклической молекулы
4.1. Построение отдельных конформаций циклогексана
1. Установите, что для расчета будет использован метод молекулярной механики, силовое поле AMBER (Setup, Molecular Mechanics, AMBER). Силовое поле содержит информацию о видах и свойствах атомов в молекуле, которая затем используется в расчетах.
2. Щелкните левой клавишей мыши на команду Options (Опции) для открытия диалогового окна Force Field Options (Опции силового поля).
3. Во вкладке Dielectric (Диэлектрическая проницаемость) выберите команду Distance Dependent (Зависит от расстояния).
Если влияние водной среды не учитывается в расчете молекулярного силового поля, следует использовать диэлектрическую постоянную, значение которой зависит от расстояния между атомами. Это уменьшит силу Кулоновского взаимодействия до 1/r2 вместо 1/r и, таким образом, будет выступать аналогом растворителя. Также Кулоновские взаимодействия можно учесть, задав коэффициент Scale Factor отличный от единицы.
4. Задайте коэффициент Scale Factor равный 1. В окне 1-4 Scale Factor (Коэффициент 1-4) задайте коэффициенты Electrostatic (электростатического взаимодействия) и van der Waals (Ван-дер-ваальсового взаимодействия), равные 0.5 каждый. Во вкладке Cutoffs (Отсечение) выберите команду None (Отсутствует). Нажмите кнопку ОК для закрытия обоих окон.
Таким образом, взаимодействием двух атомов, между которыми встроены два других атома, пренебрегают. Параметры силового поля AMBER задаются двумя вышеуказанными коэффициентами электростатического и Ван-дер-ваальсового взаимодействия.
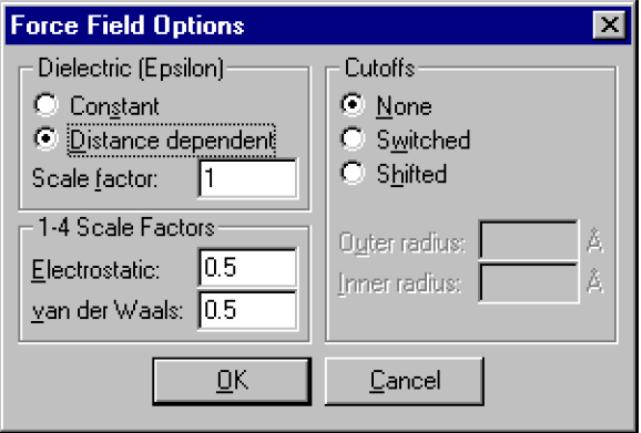
В случае с большими молекулами, взаимодействием между далеко удаленными участками можно пренебречь. Это упростит вычисления и, в таком случае, функцией Cutoffs (Отсечение) можно пренебречь.
5. В меню Setup (Установка) выберите команду Select Parameter Set (Задать параметры силового поля). В появившемся диалоговом окне выберите команду amber2 (параметры силового поля).
Программа позволяет выбрать параметры силового поля AMBER из списка или задать свои собственные. Более подробно о параметрах силового поля смотрите в руководстве программы HyperChem.
Построение конформации кресло для циклогексана
Чтобы построить конформацию кресло:
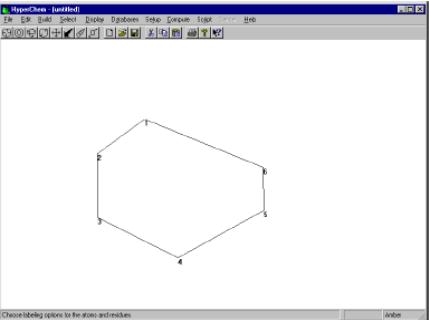
В меню Default Element (Выбрать элемент) выберите углерод и перейдите в режим рисования.
В меню Select (Выбрать) выберите команду Atoms (Атомы).
В меню Display (Отобразить) выберите команду Labels (Подписи) и пронумеруйте атомы. Для этого выберите команду Number (Нумерация).
Убедитесь, что команда Explicit Hydrogens (Показывать атомы водорода) меню Build (Построение) отключена.
Постройте молекулу, как показано на рис.
В меню Build (Построение) выберите команду Add H & Model Build (Добавить водороды и построить модель).
Отключите команду Show Hydrogens (Показывать водороды) в меню Display (Отображение).
Поворачивайте и перемещайте структуру, пока она не примет данный вид:
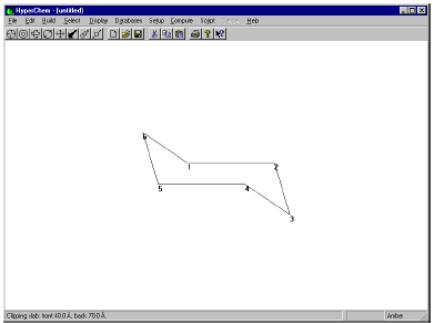
Функция Model Builder (Построение модели) позволяет построить модельную структуру. Эта структура не оптимизирована, но содержит стандартный набор длин связей, углов и торсионных углов.
Определение структурных свойств циклогексана в конформации кресло
Теперь переступим к определению структурных свойств построенной модельной структуры. Позже мы будем сравнивать эти данные с конфигурационными измерениями оптимизированной структуры.
Чтобы определить конфигурационные данные молекулы:
Откройте меню Select (Выбрать).
Выберите команду Atoms (Атомы) и отключите команду Multiple Selections (Множественный выбор).
Выделите несколько связей, углов или торсионных углов для определения конфигурации молекулы.
После выделения в строке состояния появятся следующие значения:
Длина связи: 1.54Å
Угол: 109.47°
Торсионный угол: 60°
Щелкните правой клавишей мыши на пустой части рабочего листа, чтобы снять все выделения.
Выполнение точечного расчета
Рассчитаем параметры точек и определим полную энергию неоптимизированной системы.
Чтобы провести точечный расчет:
В меню Compute (Вычислить) выберите команду Single Point (Параметры точки).
Методом точечного расчета определяют энергию в ккал/моль и суммарный среднеквадратический градиент в ккал/(моль·Å) текущей конфигурации атомов. В строке состояния появятся следующие значения:
Энергия=1.64
Градиент=3.02
В локальном минимуме суммарный среднеквадратический градиент стремится к нулю. Поэтому, согласно данных силового поля AMBER, построенная модельная структура не является локальным минимумом.
Оптимизация структуры
Приступим к минимизации конформации кресло, за счет оптимизации конформации молекулы. Сначала зададим параметры минимизации и вид минимизатора, а затем произведем вычисления.
Задание параметров оптимизации
В меню Compute (Вычислить) выберите команду Geometry Optimization (Оптимизация конформации).
Откроется следующее окно:
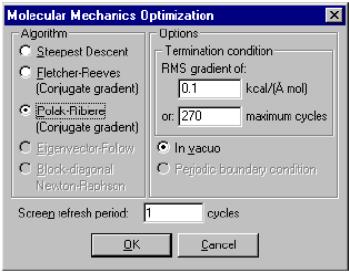
Опции данного окна позволяют задать алгоритм оптимизации и критерий сходимости для минимизации энергии.
Выберите метод сопряженных градиентов Polak-Ribiere в качестве алгоритма минимизации.
Этот алгоритм является общеупотребительным. Оба метода сопряженных градиентов (Polak-Ribiere и Fletcher-Reeves) осуществляют ряд плоскостных поисков, или циклов, в направлении сопряженных градиентов. При различных условиях используются различные методы.
Задайте среднеквадратичный коэффициент, равный 0.1. Остальные коэффициенты оставьте заданными по умолчанию.
Задавая значения среднеквадратичного градиента (RMS gradient) и максимального количества циклов (maximum cycles) Вы задаете условия для окончания вычисления. При выполнении любого из вышеуказанных условий вычисления прекращаются.
Опция In Vacuo (В вакууме) позволяет производить расчет без учета граничных условий. Это единственно-возможная опция в случае исследования системы, не имеющей молекулярного окружения. Опции Periodic Boundary (Граничные условия) и In Vacuo (В вакууме) являются взаимообратными. Невозможно выбрать опцию Periodic Boundary (Граничные условия), пока не будет выбрана опция Periodic Box (Показать молекулярное окружение).