

I.5.1. Получение алкинов.
1. Наиболее общим способом получения алкинов является дегидрогалогенирование вицинальных и геминальных дигалогенидов. Вицинальными (виц-) дигалогенидами (и, вообще, дифункциональными соединениями) называются соединения, у которых два атома галогена (или две других функциональных группы) находятся у соседних атомов углерода. Геминальными (гем-) производными называются соединения, у которых два атома галогена (или две другие функции) находятся у одного и того же атома углерода.
Дегидрогалогенирование виц- и гем-дигалогенпроизводных происходит при действии оснований:
 Х
– обычно Сl,
Br
Х
– обычно Сl,
Br
Для успешного протекания реакции необходимо использовать сильные основания (КОН, NaNH2), зачастую при нагревании до температур порядка 200 оС (более подробно реакции
отщепления будут рассмотрены позднее).
виц-Дигалогениды обычно получают путем присоединения галогенов к алкенам (см. алкены); поэтому их дегидрогалогенирование завершает переход от алкенов к алкинам; это наиболее рациональный путь такого перехода.
гем-Дигалогениды получают из альдегидов и кетонов путем замены карбонильного атома кислорода на два атома галогена; их дегидрогалогенирование завершает переход от карбонильных соединений к алкинам.
2. Также общим методом синтеза является наращивание углеродного скелета алкинов путем алкилирования ацетиленид-анионов; эта реакция будет рассмотрена ниже, при обсуждении химических свойств алкинов.
Для получения простейшего (и важнейшего) алкина – ацетилена – используются иные методы:
А. Из карбида кальция при действии воды:
СаС2 + 2 Н2О → НССН + Са(ОН)2
Б. Из метана при действии искрового разряда (~1500 оС):
2СН4 → СНСН + 3Н2
В. Из оксида углерода (II) и водорода в присутствии некоторых катализаторов (графит с интеркалированными металлами или солями):
2СО + 3Н2 → НССН + 2Н2О
I.5.2. Свойства алкинов.
По температурам кипения и плавления алкины мало отличаются от ранее рассмотренных углеводородов. Тройную связь можно обнаружить в ИК спектрах алкинов; в спектрах ЯМР 13С сигналы атомов углерода, связанных тройной связью, достаточно характеристичны.
Химические свойства алкинов, естественно, прежде всего определяются наличием связи СС. Целесообразно разделить химические реакции алкинов на две группы:
I. Реакции с разрывом тройной связи;
II. Реакции с сохранением тройной связи.
I.5.2.1. Реакции с разрывом тройной связи
Для данных реакций наблюдаются с одной стороны аналогия с реакциями двойных связей алкенов, а с другой – заметное своеобразие. Главное отличие заключается в том, что реакции электрофильного присоединения для алкинов протекают труднее, чем для алкенов, но зато для алкинов появляется некоторая способность к реакциям нуклеофильного присоединения, которое для алкенов не характерно. Причину этого можно понять, рассмотрев скоростьопределяющие (лимитирующие) стадии электрофильного и нуклеофильного присоединения к связям С=С и СС.
 Для электрофильного
присоединения:
Для электрофильного
присоединения:
Для нуклеофильного присоединения:
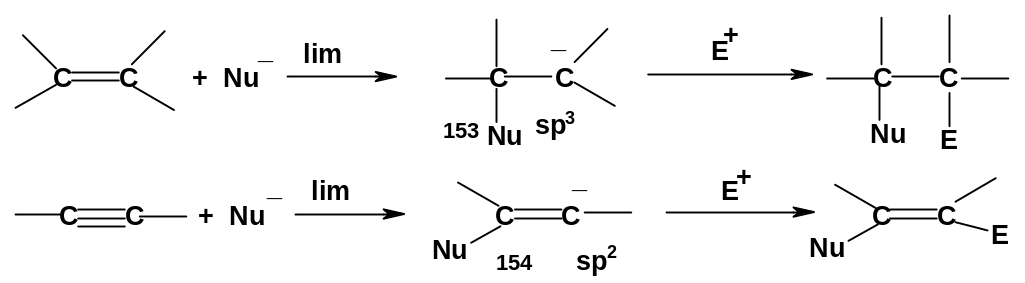 Сравним
устойчивость интермедиатов (151)-(154),
образующихся на лимитирующих стадиях
типовых реакций. Заряд в этих интермедиатах
в случае алкенов и алкинов находится
на атомах углерода, имеющих разную
степень гибридизации: в случае алкинов
орбитали атомов имеют больший s-характер.
Известно, что чем больший s-характер
имеет атом, тем выше его электроотрицательность;
электроотрицательность растет в ряду
sp3<sp2<sp.
Поэтому катионный интермедиат (152) менее
устойчив, чем катион (151), поскольку в
нем положительный
заряд находится
на более
электроотрицательном
атоме;
следовательно электрофильное
присоединение
для алкинов должно идти труднее, чем
для алкенов. Напротив, анион (154) более
устойчив, чем анион (153), что должно
облегчить протекание нуклеофильного
присоединения в алкинах по сравнению
с алкенами. В общем, данная закономерность
подтверждается экспериментальными
данными, однако не для всех реакций;
дело осложняется тем, что для некоторых
реакций механизм выяснен не до конца,
и некоторые из них трудно с полной
определенностью отнести к конкретному
типу.
Сравним
устойчивость интермедиатов (151)-(154),
образующихся на лимитирующих стадиях
типовых реакций. Заряд в этих интермедиатах
в случае алкенов и алкинов находится
на атомах углерода, имеющих разную
степень гибридизации: в случае алкинов
орбитали атомов имеют больший s-характер.
Известно, что чем больший s-характер
имеет атом, тем выше его электроотрицательность;
электроотрицательность растет в ряду
sp3<sp2<sp.
Поэтому катионный интермедиат (152) менее
устойчив, чем катион (151), поскольку в
нем положительный
заряд находится
на более
электроотрицательном
атоме;
следовательно электрофильное
присоединение
для алкинов должно идти труднее, чем
для алкенов. Напротив, анион (154) более
устойчив, чем анион (153), что должно
облегчить протекание нуклеофильного
присоединения в алкинах по сравнению
с алкенами. В общем, данная закономерность
подтверждается экспериментальными
данными, однако не для всех реакций;
дело осложняется тем, что для некоторых
реакций механизм выяснен не до конца,
и некоторые из них трудно с полной
определенностью отнести к конкретному
типу.
Реакции с разрывом тройной связи (с известной степенью условности) можно разделить на следующие типы:
А. Реакции электрофильного присоединения; Б. Реакции, катализируемые ионами металлов; В. Реакции нуклеофильного присоединения; Г. Реакции радикального присоединения; Д. Реакции олиго- и полимеризации; Е. Реакции окисления и восстановления.
А. Реакции электрофильного присоединения.
К реакциям «классического» электрофильного присоединения к алкинам относятся реакции присоединения галогенов (галогенирование) и галогеноводородов (гидрогалогенирование). К реакциям электрофильного присоединения можно отнести и гидроборирование.
Г
 алогенирование:
алогенирование:
Присоединение одного моля брома приводит к 1,2-дигалогеналкенам (155); если имеется избыток галогена, происходит дальнейшее присоединение с образованием 1,1,2,2- тетрагалогеналканов.
Присоединение галогенов к алкинам протекает медленнее, чем к алкенам, в соответствии со сформулированной выше закономерностью. Дигалогеналкены (155) в большинстве случаев имеют транс- конфигурацию, т.е. атомы брома присоединяются с разных сторон тройной связи (анти-присоединение). Это вполне аналогично галогенированию алкенов; очевидно, и механизм галогенирования алкинов аналогичен механизму галогенирования алкенов, и в качестве интермедиата образуется мостиковый ион (156):
П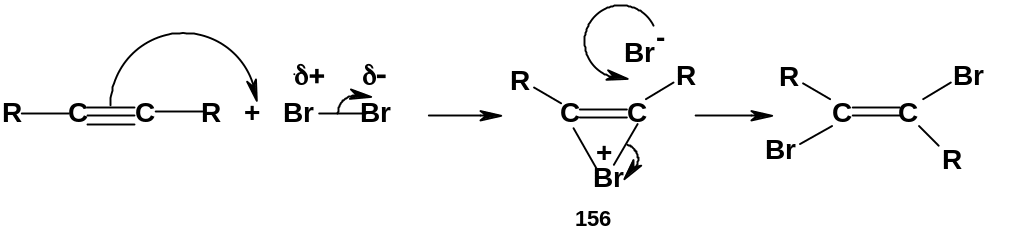 рактическое
значение имеет хлорирование самого
ацетилена (R=Н);
продукты присоединения как одного, так
и двух молей хлора используются в
качестве растворителей.
рактическое
значение имеет хлорирование самого
ацетилена (R=Н);
продукты присоединения как одного, так
и двух молей хлора используются в
качестве растворителей.
Г
 идрогалогенирование:
идрогалогенирование:
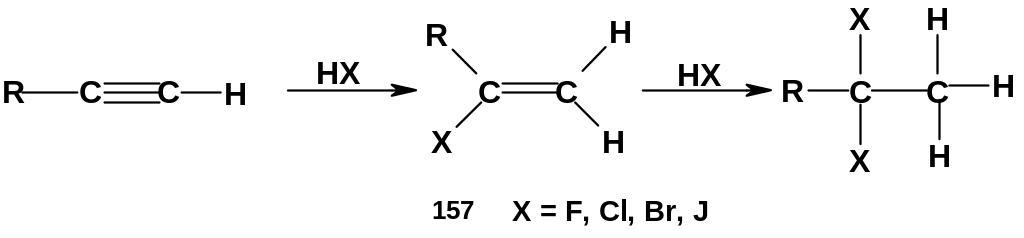
Как и в предыдущем случае, присоединение галогеноводородов идет ступенчато; присоединение одной молекулы НХ ведет к винилгалогенидам (157); присоединение второй молекулы НХ к последним приводит к гем-дигалогенидам. Присоединение галогеноводородов идет труднее, чем для алкенов: например, ацетилен присоединяет НСl только в присутствии катализаторов - солей двухвалентной ртути. Как и для предыдущей реакции, здесь наблюдается анти-присоединение к тройной связи.
Интересно проследить регионаправленность гидрогалогенирования алкинов. Присоединение одного моля галогеноводорода к терминальным алкинам (т.е. к алкинам с концевой тройной связью) идет по правилу Марковникова – как и гидрогалогенирование алкенов; это указывает на родство механизмов для обоих классов углеводородов. Присоединение второй молекулы галогеноводорода к винилгалогенидам (157) также идет по правилу Марковникова; образуются гем- (но не виц-) дигалогениды. Это требует особого объяснения.
 Рассмотрим
механизмы присоединения НХ к винилгалогениду
(157) по
правилу Марковникова (1) и против
правила Марковникова (2):
Рассмотрим
механизмы присоединения НХ к винилгалогениду
(157) по
правилу Марковникова (1) и против
правила Марковникова (2):
Механизм здесь тот же, что для простых алкенов; регионаправленность определяется устойчивостью катионного интермедиата, образующегося на скоростьопределяющей стадии присоединения протона. В варианте 1 образуется интермедиат (158), в варианте 2 – интермедиат (159). На первый взгляд может показаться, что катион (158) менее устойчив, чем катион (159), поскольку в интермедиате (158) с катионным центром связан атом галогена, обладающий сильным -I- эффектом, особенно для F и Cl (еще раз напомним – акцепторы дестабилизируют катионы). Однако именно в катионе 158 имеется сопряжение катионного центра (вакантной орбитали) с неподеленной парой электронов атома галогена, т.е атом галогена проявляет +М-эффект, приводящий к делокализации заряда [в катионе (159) сопряжения нет и +М-эффект не проявляется]. Хотя +М-эффект атомов галогена довольно слабый и по абсолютной величине уступает их –I-эффекту, решающую роль играет делокализация заряда: катион (158) более устойчив, чем катион (159), и реакция идет по правилу Марковникова.
3. Гидроборирование. Алкины присоединяют гидриды бора. В отличие от алкенов, сам боран здесь обычно не используют, т.к. получаются сложные смеси продуктов; лучшие результаты дает использование дизамещенных боранов R2ВН, где R-достаточно объёмный радикал, например циклогексил:
Э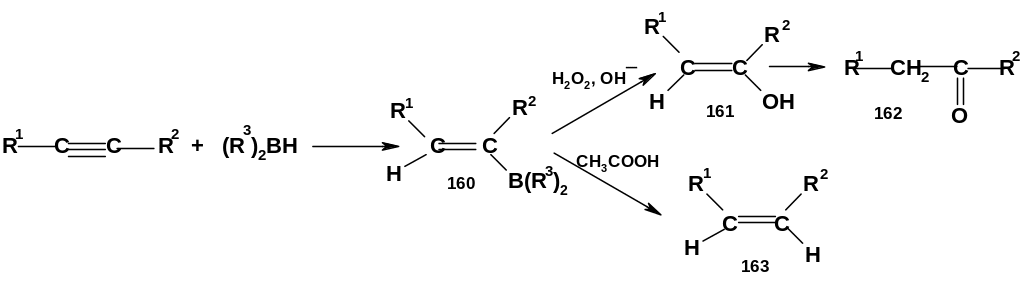 лектрофильной
частицей здесь является R2В;
как и в случае алкенов, гидроборирование
идет по схеме син-
присоединения (очевидно, через циклическое
переходное состояние); аддукт (160) имеет
цис-конфигурацию.
Если алкин терминальный (R2=Н),
фрагмент R2В
присоединяется к концевому
атому углерода. При обработке аддукта
пероксидом водорода в щелочной среде
образуется виниловый спирт (161) (енол),
который сразу перегруппировывается в
карбонильное соединение (162): при R2=Н
(терминальный алкин) образуется альдегид,
при R2Н
– кетон. Обработка аддукта (160) водным
раствором уксусной кислоты приводит к
цис-алкену (163).
лектрофильной
частицей здесь является R2В;
как и в случае алкенов, гидроборирование
идет по схеме син-
присоединения (очевидно, через циклическое
переходное состояние); аддукт (160) имеет
цис-конфигурацию.
Если алкин терминальный (R2=Н),
фрагмент R2В
присоединяется к концевому
атому углерода. При обработке аддукта
пероксидом водорода в щелочной среде
образуется виниловый спирт (161) (енол),
который сразу перегруппировывается в
карбонильное соединение (162): при R2=Н
(терминальный алкин) образуется альдегид,
при R2Н
– кетон. Обработка аддукта (160) водным
раствором уксусной кислоты приводит к
цис-алкену (163).
Б. Реакции, катализируемые ионами металлов.
Ряд важных реакций алкинов катализируется ионами Hg2+ и Сu+. Из подобных реакций наиболее известна реакция гидратации самого ацетилена и его гомологов. Гидратацию алкинов, в принципе, можно проводить просто в кислой среде (как гидратацию алкенов); однако в этих условиях алкины присоединяют воду заметно труднее, чем алкены. Реакция заметно ускоряется в присутствии ионов Hg2+; впервые это было установлено на примере ацетилена М.Г.Кучеровым (реакция Кучерова).
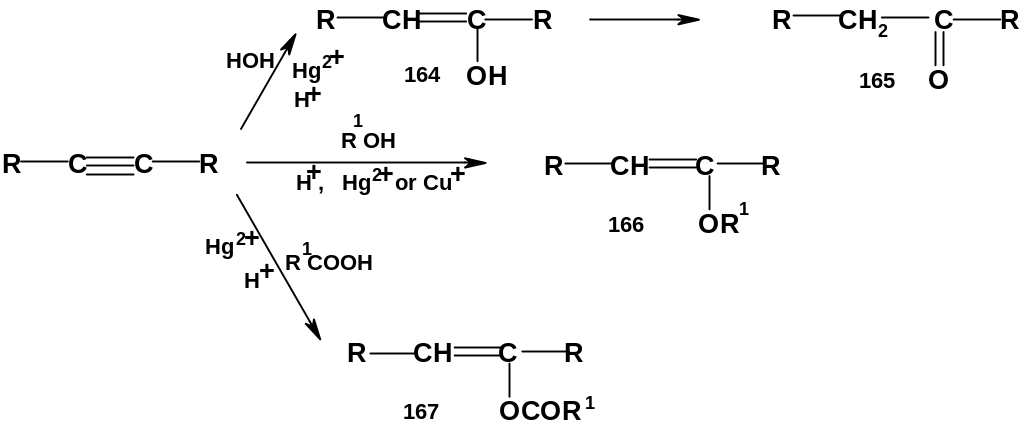 Присоединение
воды приводит к енолам (164), которые
перегруппировываются в карбонильные
соединения (165) (аналогичная ситуация
приводилась выше при рассмотрении
гидроборирования алкинов). Здесь альдегид
(уксусный) получается только из ацетилена
(R=Н);
именно получение уксусного альдегида
из ацетилена и было впервые изучено
М.Г.Кучеровым.
Во всех остальных случаях образуются
кетоны, в частности, терминальные алкины
R-CCH
образуют метилкетоны R-CO-CH3;
этот результат противоположен результату
гидроборирования.
Присоединение
воды приводит к енолам (164), которые
перегруппировываются в карбонильные
соединения (165) (аналогичная ситуация
приводилась выше при рассмотрении
гидроборирования алкинов). Здесь альдегид
(уксусный) получается только из ацетилена
(R=Н);
именно получение уксусного альдегида
из ацетилена и было впервые изучено
М.Г.Кучеровым.
Во всех остальных случаях образуются
кетоны, в частности, терминальные алкины
R-CCH
образуют метилкетоны R-CO-CH3;
этот результат противоположен результату
гидроборирования.
В присутствии ионов Hg2+ или Cu+ в кислой среде алкины присоединяют спирты с образованием простых виниловых эфиров (простых эфиров енолов) (166); карбоновые кислоты присоединяются к алкинам в присутствии ионов Hg2+ с образованием сложных эфиров енолов (167). Продукт присоединения уксусной кислоты к ацетилену – винилацетат [(167), R=H, R1=CH3)] – является мономером для получения важнейших полимеров – поливинилацетата и поливинилового спирта.
Механизмы реакций, катализируемых ионами Hg2+ и Cu+, во многом не выяснены. Вероятнее всего, вначале ион металла образует π-комплекс с тройной связью алкина; относительно дальнейшего протекания реакции есть разные мнения. Согласно одной версии, скоростьопределяющей стадией является нуклеофильная атака молекулы воды (спирта); тогда реакцию следует относить к нуклеофильному присоединению. Согласно другой версии лимитирующей стадией является присоединение протона, т.е. электрофильная атака; в этом случае реакция является электрофильным присоединением. Поскольку полной ясности тут нет, логично выделить эти реакции в отдельную группу.
В. Реакции нуклеофильного присоединения.
Наиболее известной реакцией присоединения к связи СС, протекающей по чисто нуклеофильному механизму, является присоединение спиртов при катализе сильными основаниями. Например, ацетилен присоединяет спирты при нагревании в присутствии твердого КОН под давлением:
П![]() родуктами
реакции являются виниловые эфиры
(простые эфиры, содержащие винильный
радикал); если посмотреть на результат
реакции «со стороны спирта», то легко
заметить, что в итоге реакции атом
водорода в спирте замещается на винильную
группу; поэтому данную реакцию иногда
называют винилированием
спиртов.
родуктами
реакции являются виниловые эфиры
(простые эфиры, содержащие винильный
радикал); если посмотреть на результат
реакции «со стороны спирта», то легко
заметить, что в итоге реакции атом
водорода в спирте замещается на винильную
группу; поэтому данную реакцию иногда
называют винилированием
спиртов.
 Механизм реакции
следующий:
Механизм реакции
следующий:
Сильное основание (в данном случае гидроксид-анион) отрывает протон от молекулы спирта, генерируя алкоксид-анион RO-; алкоксид-анион, являясь сильным нуклеофилом, присоединяется к тройной связи ацетилена; при этом образуется карбанионный интермедиат (168); этот интермедиат стабилизируется путем отрыва протона от молекулы спирта. Стадия присоединения алкоксид-аниона является скоростьопределяющей (лимитирующей); именно поэтому присоединение является нуклеофильным [еще раз подчеркнем – в ранее рассмотренных реакциях электрофильного присоединения к связям С=С и СС при протекании лимитирующей стадии реагент –например, протон - являлся акцептором электронной пары, в данном же случае реагент – алкоксид-анион – является донором электронной пары].
Аналогично спиртам к связи СС могут присоединяться тиолы RSH.
Г. Реакции радикального присоединения.
Данные реакции аналогичны соответствующим реакциям присоединения к алкенам; в частности, известно «антимарковниковское» присоединение НBr к монозамещенным ацетиленам RССН в присутствии пероксидов и других инициаторов радикальных реакций.
Д. Реакции олиго- и полимеризации.
Реакции олигомеризации весьма характерны для алкинов, в первую очередь для самого ацетилена; данные реакции идут по различным схемам и в различных условиях, чаще всего при катализе соединениями металлов.
1. Линейная димеризация ацетилена
![]() Данная
реакция проходит в кислых средах в
присутствии ионов Сu+:
Данная
реакция проходит в кислых средах в
присутствии ионов Сu+:
Происходит присоединение одной молекулы ацетилена к другой; продуктом реакции является винилацетилен, который находит применение в органическом синтезе.
Циклоолигомеризация ацетилена и его производных.
Данные превращения относятся к реакциям циклоприсоединения.
 2а. Циклотримеризация.
2а. Циклотримеризация.
В данной реакции образуются производные бензола; из самого ацетилена (R=Н) образуется бензол. Реакция может идти: а. при нагревании; б. в присутствии конц. Н2SO4; в. лучше всего – при металлокомплексном катализе, например, при использовании Ni(CO)2. (Ph3P)2.
Реакцию можно отнести к [2+2+2]- циклоприсоединению.
 2б.
Циклотетрамеризация
2б.
Циклотетрамеризация
При катализе цианидом никеля под давлением четыре молекулы ацетилена образуют соединение с восьмичленным циклом – циклооктатетраен. Такая достаточно редкая реакция становится возможной благодаря тому, что предварительно образуется комплекс четырех молекул ацетилена с ионом никеля; в этом комплексе молекулы ацетилена занимают такое пространственное положение, что получается «заготовка» для восьмичленного цикла. Данную реакцию можно считать [2+2+2+2]-циклоприсоединением.
![]() Полимеризация
алкинов протекает в присутствии
инициаторов радикальных реакций или
металлорганических катализаторов:
Полимеризация
алкинов протекает в присутствии
инициаторов радикальных реакций или
металлорганических катализаторов:
Полученные полимеры содержат систему сопряженных двойных связей и представляют интерес как органические полупроводники.
Е. Реакции окисления и восстановления.
Реакции окисления алкинов, затрагивающие тройную связь, имеют несколько меньшее значение, чем соответствующие реакции алкенов. При действии на дизамещенные ацетилены сильных окислителей типа перманганата или бихромата калия возможны два основных результата:
Б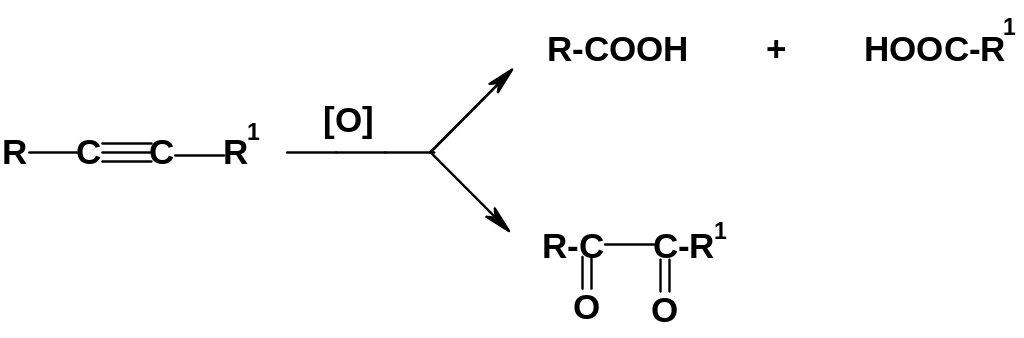 олее
общий характер имеет первый вариант –
полный разрыв тройной связи с образованием
двух карбоновых кислот; результат вполне
аналогичен соответствующей реакции
алкенов. Значительно интереснее второй
вариант – неполное окисление с
образованием 1,2-дикетонов; эти соединения
перспективны для использования в
органическом синтезе. Однако такой
результат достигнут лишь в отдельных
случаях; другие способы получения
1,2-дикетонов в настоящее время более
«конкурентоспособны».
олее
общий характер имеет первый вариант –
полный разрыв тройной связи с образованием
двух карбоновых кислот; результат вполне
аналогичен соответствующей реакции
алкенов. Значительно интереснее второй
вариант – неполное окисление с
образованием 1,2-дикетонов; эти соединения
перспективны для использования в
органическом синтезе. Однако такой
результат достигнут лишь в отдельных
случаях; другие способы получения
1,2-дикетонов в настоящее время более
«конкурентоспособны».
Восстановление связи СС алкинов ведет к алкенам и далее к алканам. Наиболее интересны варианты, при которых реакция останавливается на стадии получения алкенов. При восстановлении дизамещенных ацетиленов можно направить реакцию в сторону получения как цис-, так и транс-алкенов:
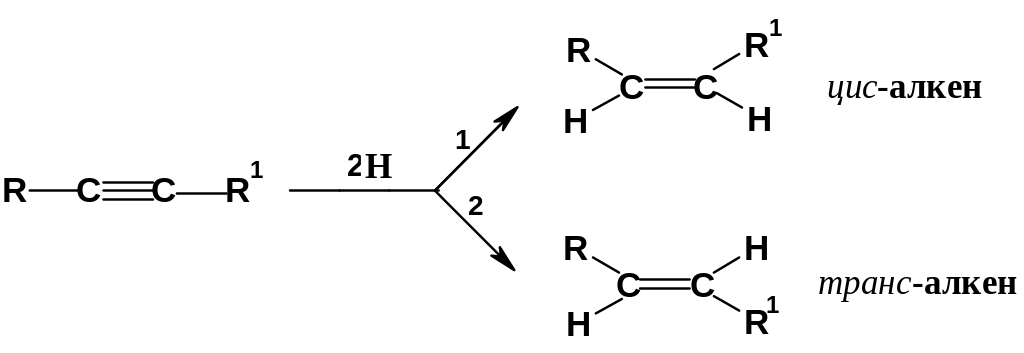 цис-Алкены
образуются: А. При восстановлении
водородом в присутствии катализатора
– палладия, нанесенного на карбонат
кальция (катализатор Линдлара);
Б. При восстановлении специфическим
реагентом – диимидом (169); этот неустойчивый
реагент образуется при окислении
гидразина солями Сu2+
и действует «в момент образования» без
выделения в чистом виде; диимид при этом
окисляется до азота:
цис-Алкены
образуются: А. При восстановлении
водородом в присутствии катализатора
– палладия, нанесенного на карбонат
кальция (катализатор Линдлара);
Б. При восстановлении специфическим
реагентом – диимидом (169); этот неустойчивый
реагент образуется при окислении
гидразина солями Сu2+
и действует «в момент образования» без
выделения в чистом виде; диимид при этом
окисляется до азота:
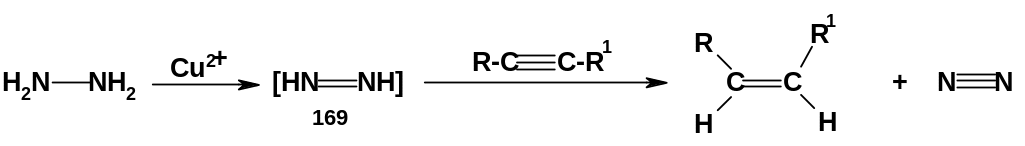
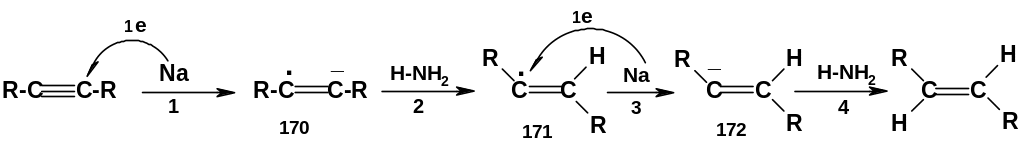 транс-Алкены
образуются при восстановлении дизамещенных
ацетиленов натрием (или литием) в жидком
аммиаке. Предполагается, что эта реакция
протекает по
ион-радикальному
механизму,
т.е. с образованием в качестве интермедиатов
ион-радикалов:
транс-Алкены
образуются при восстановлении дизамещенных
ацетиленов натрием (или литием) в жидком
аммиаке. Предполагается, что эта реакция
протекает по
ион-радикальному
механизму,
т.е. с образованием в качестве интермедиатов
ион-радикалов:
Вначале атом натрия отдает алкину один электрон (стадия 1); образуется анион-радикал (170) [ еще раз напомним: если к нейтральной молекуле присоединяется один электрон, образуется анион-радикал, а если она теряет один электрон – то катион-радикал]. Далее карбанионный центр ион-радикала отрывает протон от молекулы аммиака (стадия 2); образуется радикал (171); этот радикал принимает один электрон от следующего атома натрия (стадия 3) с образованием карбаниона (172); наконец, карбанион отрывает протон от очередной молекулы аммиака с образованием конечного продукта – транс-алкена. По всей видимости, лимитирующей стадией является стадия 2; возникающий при этом радикал (171) имеет транс-конфигурацию – она более устойчива, чем цис-конфигурация; это и приводит к образованию именно транс-алкена.