

62.Биоокисление жирных кислот
Окисление жирных кислот в организмах - чрезвычайно важный процесс, он может протекать по α-, β- и ω-углеродным атомам жирных кислот. Основной путь окисления жирных кислот как в животных, так и в растительных тканях - это β-окисление.
β-Окисление жирных кислот.β-Окисление жирных кислот было впервые изучено в 1904 г. Ф. Кноопом. В дальнейшем было установлено, что β- окисление осуществляется только в митохондриях. Благодаря работам Ф. Линена с сотрудниками (1954-1958 гг.) были выяснены основные ферментативные процессы окисления жирных кислот. В честь ученых, открывших данный путь окисления жирных кислот, процесс β-окисление получил название цикла Кноопа-Линена.
По современным представлениям, процессу окисления жирных кислот предшествует их активация в цитоплазме с участием ацил-КоА-синтетазыи с использованием энергии АТФ:
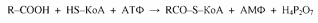
В форме ацил-КоА жирные кислоты поступают в митохондрии, в матриксе которых они подвергаются β-окислению, включающему последовательность нижеприведенных ферментативных окислительно-восстановительных реакций.
Первой реакцией
на пути расщепления жирных кислот
является дегидрирование с образованием
транс-2,3-ненасыщенных производных,
катализируемое различными ФАД-содержащими
ацил-КоА-дегидрогеназами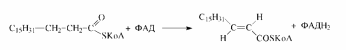 Вторая
реакция - гидратация двойной связи -
катализируется еноил-КоА - гидратазой:
Вторая
реакция - гидратация двойной связи -
катализируется еноил-КоА - гидратазой:
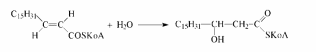
На следующей (третьей) стадии происходит дегидрирование спиртового фрагмента, которое осуществляется соответствующей дегидрогеназой и окисленной формой кофермента НАД:
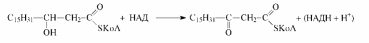
В результате окисления образуется β-оксокислота, из-за чего весь процесс в целом и получил название β-окисления.
Четвертая,
последняя реакция, катализируемая
тиолазой, сопровождается
окислительно-восстановительным
расщеплением связи Сα-Сβ с отщеплением
ацетил-КоА и присоединением остатка
КоА по месту разрыва межуглеродной
связи:
Эта реакция носит название тиолиза и является высоко экзергонической, поэтому равновесие в ней всегда смещено в сторону образования продуктов.
Последовательное повторение этого цикла реакций приводит к полному распаду жирных кислот с четным числом атомов углерода до ацетил-КоА. В результате этого процесса образуются ацетил-КоА, ФАДН2 и НАД-Н. Далее ацетил-КоА вступает в цикл Кребса, а восстановленные коферменты - в дыхательную цепь.
Особенности окисления жирных кислот с нечетным числом углеродных атомов заключается в том, что наряду с обычными продуктами окисления, образуется одна молекула СН3-СН2-СО~SКоА (пропионил-КоА), которая в процессе карбоксилирования переводится в сукцинил-КоА, поступающий в цикл Кребса.
Особенности окисления ненасыщенных жирных кислот определяются положением и числом двойных связей в их молекулах. До места двойной связи ненасыщенные жирные кислоты окисляются так же, как и насыщенные. Если двойная связь имеет ту же транс-конфигурацию и расположение, что и еноил-КоА, то далее окисление идет по обычному пути. В противном случае в реакциях участвует дополнительный фермент, который перемещает двойную связь в нужное положение и изменяет конфигурацию молекулы кислоты.
При β-окислении жирных кислот выделяется большое количество энергии. При полном окислении одного моля жирной кислоты, содержащей 2n атомов углерода, образуется n молей ацетил-КоА и (n-1) молей (ФАДН2 + НАДН). Окисление ФАДН2 дает 2АТФ, а при окислении НАДН образуется 3АТФ. Полное сгорание одного моля ацетил-КоА приводит к образованию 12 молей АТФ.
Окисление жирных кислот с четным числом углеродных атомов.
Жирные кислоты, входящие в состав естественных жиров животных и растений, имеют четное число углеродных атомов. Любая такая кислота, от которой отщепляется по паре углеродных атомов, в конце концовпроходит через стадию масляной кислоты. После очередного β-окисления масляная кислота становится ацетоуксусной. Последняя затем гидролизуется до двух молекул уксусной кислоты.
Окисление жирных кислот с нечетным числом углеродных атомов.
Как отмечалось, основная масса природных липидов содержит жирные кислоты с четным числом углеродных атомов. Однако в липидах многих растений и некоторых морских организмов присутствуют жирные кислоты с нечетным числом атомов углерода. Кроме того, у жвачных животных при
переваривании углеводов в рубце образуется большое количество пропионовой кислоты, которая содержит три углеродных атома. Пропионатвсасывается в кровь и окисляется в печени и других тканях. Установлено,
что жирные кислоты с нечетным числом углеродных атомов окисляютсятаким же образом, как и жирные кислоты с четным числом углеродных атомов, с той лишь разницей, что на последнем этапе расщепления
(β-окисления) образуется одна молекула пропионил-КоА и одна молекула ацетил-КоА, а не 2 молекулы ацетил-КоА. Активированный трехуглеродный фрагмент – пропионил-КоА – включается в цикл трикарбоновых кислот после превращения в сукцинил-КоА.
болезни связанные с нарушением окисления жирных кислот.
Изучение митохондриальныхболезней, обусловленных нарушением бета окисления жирных кислот с различной длиной углеродной цепи было начато в 1976 г., когда учёные впервые описали больных с дефицитом ацил-КоА-дегидрогеназысреднецепочечных жирных кислот и глутаровойацидемией II типа. В настоящее время эта группа заболеваний включает не менее 12 самостоятельных нозологических форм, происхождение которых связано с генетически детерминированными расстройствами трансмембранного транспорта жирных кислот (системный дефицит карнитина, дефицит карнитинпальмитоилтрансфераз I и II, ацилкарнитин-карнитинтранслоказы) и их последующего митохондриального бета-окисления (дефицит ацил-КоА и 3-гидрокси-ацил-КоА-дегидрогеназ жирных кислот с различной длиной углеродной цепи, глутароваяацидемия II типа). Частота дефицита ацил-КоАдегидрогеназысреднецепочечных жирных кислот составляет 1:8900 новорождённых, частота других форм патологии пока не установлена.
Генетические данные и патогенез. Заболевания имеют аутосомно-рецессивный тип наследования.
Патогенез болезней обмена жирных кислот связан с истощением углеводных запасов в условиях метаболического стресса (интеркуррентных инфекционных болезней, физической или эмоциональной перегрузке, голодании, хирургическом вмешательстве). В подобной ситуации липиды становятся необходимым источником восполнения энергетических потребностей организма. Происходит активация дефектных процессов транспорта и бета-окисления жирных кислот. Вследствие мобилизации омега-окисления происходит накопление в биологических жидкостях дикарбоновых кислот, их токсичных производных, конъюгатов карнитина - в результате развивается вторичная карнитиновая недостаточность.
Симптомы. Клинические проявления всех болезней обмена жирных кислот имеют большое сходство. Заболевания, как правило, характеризуются приступообразным течением. Существуют тяжёлая (ранняя, генерализованная) и лёгкая (поздняя, мышечная) формы, отличающиеся разной степенью ферментного дефицита или его тканевой локализацией.
Тяжёлая форма манифестирует в раннем возрасте, в том числе в периоде новорождённости. Основные симптомы: рвота, генерализованные тонико-клонические судороги или инфантильные спазмы, прогрессирующие вялость, сонливость, общая мышечная гипотония, нарушение сознания вплоть до комы, расстройство сердечной деятельности (нарушение ритма или кардиомиопатия), увеличение печени (синдром Рея). Заболевание сопровождается летальностью (до 20%) и риском внезапной детской смерти.
Лёгкая форма обычно впервые проявляется в школьном возрасте и у подростков. Развиваются боли в мышцах, слабость, утомляемость, моторная неловкость, тёмная окраска мочи (миоглобинурия).
Характерные дополнительные клинические признаки дефицита 3-гидрокси-ацил-Ко А-дегидрогеназы жирных кислот с длинной углеродной цепью - периферическаянейропатия и пигментный ретинит. У будущих матерей, дети которых, вероятно, будут иметь этот ферментный дефект, нередко осложняется течение беременности - развиваются жировая инфильтрация печени, тромбоцитопения, повышается активность трансаминаз.
63.биосинтез жирных кислот. Особенности синтеза ненасыщенных жирных кислот. Незаменимые жирные кислоты. Синтез длинноцепочечных насыщенных и ненасыщенных жирных кислот.
БИОСИНТЕЗ ЖИРНЫХ КИСЛОТ
Биосинтез жирных кислот и липидов играет важную роль в жизнедеятельности организмов. Именно в виде жирных кислот и триацилглицеринов откладываются основные количества энергетических ресурсов организмов животных, в то время как энергоресурсы, откладываемые в форме углеводов, незначительны.
В клетках организма жирные кислоты синтезируются из ацетил-КоА, образующегося из избыточной глюкозы пищи, которая не была использована организмом на энергетические нужды. В качестве восстановителя в биосинтезе жирных кислот принимает участие НАДФН, синтезируемый, в основном, в пентофосфатном пути распада углеводов. Нужно отметить, что хотя все реакции β-окисления жирных кислот обратимы, этот путь не используется организмом с целью их синтеза. Биосинтез жирных кислот осуществляется в цитоплазме клеток и катализируется целым полиферментным надмолекулярным ансамблем - пальмитилсинтетазой, состоящей из семи ферментов.
Суммарная реакция биосинтеза жирных кислот в цитоплазме имеет следующий вид (Е - пальмитилсинтетаза):
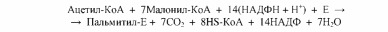
Из данного уравнения можно видеть, что для синтеза жирной кислоты требуется всего одна молекула ацетил-КоА, служащая «затравкой». Непосредственным источником синтеза является малонил-КоА, который образуется из ацетил-КоА по реакции:
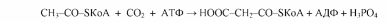
Эта реакция катализируется биотинзависимым ферментом - ацетил-КоА- карбоксилазой. Функция биотина сводится к переносу диоксида углерода на субстрат.
Пальмитилсинтетаза представляет собой многофункциональный ансамбль белков: в центре полиферментного ансамбля находится ацилпереносящий белок (АПБ), содержащий свободную SH-группу; шесть остальных ферментов располагаются по периметру, причем один из них также содержит SH- группу.
Процесс синтеза жирной кислоты описывается рядом последовательных реакций:
1. Перенос
ацетила с ацетил-КоА на синтетазу: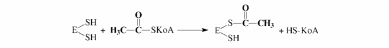
2. Перенос
малонила с малонил-КоА на синтетазу:
3. Конденсация ацетила с малонилом и декарбоксилирование образовавшегося продукта:
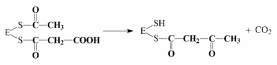
4. Первое восстановление промежуточного продукта с участием НАДФН:
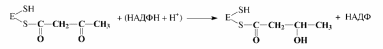
5. Дегидратация
промежуточного продукта: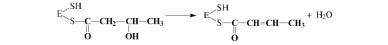
6. Второе
восстановление промежуточного продукта
с участием НАДФН:
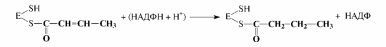
Затем синтезированный бутирил перемещается на ту SH-группу, с кото¬рой был связан затравочный ацетил, а на освободившуюся SH-группу поступа¬ет новый малонильный остаток из малонил-КоА. Далее цикл повторяется снова; после семи оборотов цикла синтезируется пальмитил-Е, который при участии пальмитилдеацилазыгидролизуется до пальмитиновой кислоты и фермента (Е). Пальмитиновая кислота - это основной продукт биосинтеза, однако в неболь¬ших количествах могут образовываться и другие жирные кислоты.
Жирные кислоты с разветвленной углеродной цепью синтезируются из продуктов метаболизма аминокислот с разветвленной цепью (валин, изолейцин и лейцин) через ацильные производные КоА путем удлинения цепи и при уча¬стии АПБ. Особенности биосинтеза полиненасыщенных жирных кислот пред¬ставляют интерес в связи с их витаминоподобными функциями. Некоторые полиеновые кислоты могут синтезироваться из олеино¬вой кислоты с помощью ряда последовательных реакций. Однако, синтез полиненасыщенных кислот, содержащих двойные связи, расположенные между ко¬нечным метилом и седьмым атомом углерода, невозможен, поэтому они и яв¬ляются незаменимыми в пищевом рационе.
Таким образом, биосинтез и поступление с пищей - два основных источ¬ника жирных кислот для организма человека и животных.
Образование ненасыщенных жирных кислот.
В отличие от растительных тканей ткани животных обладают весьма ограниченной способностью превращать насыщенные жирные кислоты в ненасыщенные. Установлено, что две наиболее распространенные мононенасыщенные жирные кислоты – пальмитоолеиновая и олеиновая – синтезируются из пальмитиновой и стеариновой кислот.Эти превращения протекают в микросомах клеток печени и жировой ткани при участии молекулярного кислорода, восстановленной системы пиридиновых нуклеотидов и цитохрома b5. Превращению подвергаются только активированные формы пальмитиновой и стеариновой кислот.
Ферменты, участвующие в этих превращениях, получили название десатураз. Наряду с десатурацией жирных кислот (образование двойных связей) в микросомах происходит и их удлинение (элонгация), причем оба эти процесса могут сочетаться и повторяться. Удлинение цепи жирной кислотыпроисходит путем последовательного присоединения к соответствующемуацил-КоАдвууглеродных фрагментов при участии малонил-КоА и НАДФН. Энзиматическая система, катализирующая удлинение жирных кислот, получила название элонгазы. На схеме представлены пути превращения пальмитиновой кислоты в реакциях десатурации и элонгации.
Незаменимые жирные кислоты.
В настоящее время показано, что в микросомах клеток млекопитающих образование двойных связей может происходить только на участке цепи жирной кислоты от 9-го до 1-го углеродных атомов, ибо в микросомах
отсутствуют десатуразы, которые могли бы катализировать образование двойных связей в цепи далее 9-го углеродного атома. У животных двойные связи могут образовываться в Δ4-, Δ5-, Δ6- и Δ9-положении, но не далее Δ9-положения, в то время как у растений – в Δ6-, Δ9-, Δ12 и Δ15-положении.
Поэтому в организме млекопитающих, в том числе и человека, не могут образовываться, например, из стеариновой кислоты (18:0) линолевая (18:2; 9,12) и линоленовая (18:3; 9,12,15) кислоты. Эти кислоты относятся к категории незаменимых жирных кислот. К незаменимым жирным кислотам обычно относят также арахидоновую кислоту (20:4; 5,8,11,14). У большинства млекопитающих арахидоноваякислота может образовываться из линолевой кислоты. Незаменимые жирные кислоты должны поступать в организм с пищей.
При длительном их отсутствии в пище у животных наблюдается отставание в росте, развиваются характерные поражения кожи и волосяного покрова. Описаны случаи недостаточности незаменимых жирных кислот и у человека. Так, у детей грудного возраста, получающих искусственное питание
с незначительным содержанием жиров, может развиться чешуйчатый дерматит, который поддается лечению препаратом линолевой кислоты. Нарушения, обусловленные недостатком незаменимых жирных кислот, наблюдаются также у больных, жизнедеятельность которых в течение длительного времени поддерживается только за счет внутривенного питания, почти лишенного жирных кислот. Принято считать, что во избежание этих нарушений необходимо, чтобы на долю незаменимых жирных кислот приходилось не менее 1–2% от общей потребности в калориях. Следует отметить, что незаменимые жирные кислоты содержатся в достаточнобольших количествах в растительных маслах.
Исследования, проведенные с применением изотопов, показали, что арахидоновая кислота и некоторые другие 20-углеродные (эйкозановые) кислоты, содержащие двойные связи, участвуют в образовании эйкозаноидов.
64. метаболизм сложных липидов. Наследственные болезни, связанные с нарушением катаболизма сложных липидов.
МЕТАБОЛИЗМ ФОСФОЛИПИДОВ
В отличие от триглицеридов и жирных кислот фосфолипиды не являются существенным энергетическим материалом. Фосфолипиды играют важную роль в структуре и функции клеточных мембран, активации мембранныхи лизосомальных ферментов, в проведении нервных импульсов, свертывании крови, иммунологических реакциях, процессах клеточной пролиферации и регенерации тканей, в переносе электронов в цепи «дыхательных» ферментов. Особая роль фосфолипидам отводится в формировании липопротеидных комплексов.
Биосинтез фосфолипидов интенсивно происходит в печени, стенке кишечника, семенниках, яичниках, молочной железе и других тканях. Наиболее важные фосфолипиды синтезируются главным образом в эндоплазматической сети клетки.
Центральную роль в биосинтезе фосфолипидов играют 1,2-диглицериды (в синтезе фосфатидилхолинов и фосфатидилэтаноламинов), фосфатиднаякислота (в синтезе фосфатидилинозитов) и сфингозин (в синтезе сфингомиелинов). Цитидинтрифосфат (ЦТФ) участвует в синтезе практически всех фосфолипидов. В качестве примера рассмотрим синтез отдельных представителей фосфолипидов.
Гликолипидозы – группа заболеваний, обусловленных нарушением распада гликолипидов (жироподобные вещества, содержащие углеводы).
Наследственные болезни, связанные с нарушением катаболизма сложных липидов.
Гликолипидозы в основном наследуются по аутосомно-рецессивному типу, за исключением синдрома Фабри, дефект при котором связан с Х-хромосомой.
Гликолипидозы подразделяются на:
цереброзидозы (болезнь Гоше, болезнь Краббе);
сульфатидозы (метахроматическая лейкодистрофия);
церамидолигозидозы (церамидлактозидлипоидоз, синдром Фабри);
ганглиозидозы (От2-ганглиозидозы: болезнь Тея – Сакса, болезнь Зандгоффа – Яцкевича – Пильца, болезнь Бернгеймера – Зайтельберга, инфантильный ганглиозидоз ГУ типа; От,-ганглиозидозы: болезнь Норманна – Ландинга, болезнь Дерри).
В основе болезни Гоше лежит утрата активности одного из ферментов, которая приводит к накоплению в клетках физиологической защитной системы организма глюкоцереброзидов.
Выделяются три типа болезни:
детский (инфантильный) тип с выраженными расстройствами нервной системы (повышение тонуса мышц, приводящее в поздних стадиях к сокращению мышц спины и шеи с запрокидыванием головы и вытягиванием конечностей, неритмичным судорогам в течение нескольких минут, судорогам жевательных мышц), развивается в возрасте около 6 месяцев;
хронический или тип 1 с нарушениями со стороны нервной системы;
юношеский тип, по тяжести течения занимает промежуточное положение.
Основными симптомами заболевания являются отставание в физическом развитии, инфантилизм, увеличение печени и селезенки, последняя достигает огромных размеров, занимая почти весь живот. Почти у всех больных отмечаются проявления гиперспленизма в виде анемии, лейкопении, тромбоцитопении. Наблюдаются также склонность к кровотечениям, кожные геморрагические высыпания, обильные маточные кровотечения.
В основе болезни Тея – Сакса лежит нарушение обмена ганглиозидов (природные органически соединения в тканях организма), сопровождающееся повышением их уровня в мозге в 100-300 раз, также в печени, селезенке. Тип наследования аутосомно-рецессивный. Частота заболевания 1 случай на 250 тыс. человек, среди евреев-ашкенази – 1 на 4 тыс.
При рождении и в первые 3-4 месяца жизни дети не отличаются от здоровых сверстников. Заболевание развивается очень медленно. Постепенно ребенок становится менее активным, теряет приобретенные навыки, утрачивает интерес к игрушкам.
С 4-6 месяцев начинают развиваться психомоторные нарушения. Дети становятся апатичными, перестают интересоваться окружающим, наблюдается мышечная гипотония.
К концу 1-го года жизни развивается слепота, обусловленная атрофией зрительных нервов. Интеллект снижается до уровня идиотии. Постепенно развивается полная обездвиженность, наблюдаются судороги, не поддающиеся терапии.
Смерть обычно наступает в возрасте 2-4 года.
Сфингомиелиноз или болезнь Ниманна – Пика – это внутриклеточный липидоз, характеризующийся накоплением в клетках, которые несут функцию физиологической защиты организма, фосфолипида сфингомиелина из-за нарушения активности сфингомиелиназы. Тип наследования аутосомно-рецессивный. Частота заболевания составляет примерно 1 случай на 10 тыс. человек.
Заболевание встречается только в раннем детском возрасте и характеризуется злокачественным течением. Сначала проявляются отказ от еды, срыгивания и рвота. Затем начинается задержка психомоторного развития, прогрессирует увеличение печени и селезенки. В дальнейшем развиваются ослабление произвольных движений рук и ног, глухота, слепота. Наблюдается увеличение лимфатических узлов. Кожа приобретает коричневый оттенок. На глазном дне обнаруживается вишнево-красное пятно.
Выделяются 4 варианта болезни – А, В, С, D. При вариантах А и В отмечается дефицит сфингомиелиназы, при вариантах С и D активность энзима нормальная.
При варианте болезни А (классическая форма) имеются тяжелые поражения нервной системы, которые сочетаются с выраженным увеличение печени и селезенки.
Вариант В отличается более поздними сроками проявления (2 - 6 лет), отсутствием неврологической симптоматики и хроническим течением.
Чрезвычайной вариабельностью начала характеризуется вариант С. У большинства (40%) больных отмечается длительная желтуха, исчезающая к 2-4 месяцам жизни, типично увеличение печени и селезенки.
Вариант D по клинической картине сходен с вариантом С, у больных часто наблюдаются импульсивные малые судороги.
Гиперлипопротеинемии обусловлены нарушениями обмена липидов крови вследствие дефектов ферментов или клеточных рецепторов.
Описано пять отдельных подтипов первичнойгиперлипидемии (I, II, III, IV и V).
Наиболее распространенная форма наследуемойгиперлипидемии детского возраста - семейная гиперхолестеринемия. Частота ее встречаемости – 1 случай среди 500 здоровых людей.
При ней выявляется повышение холестерина при нормальном содержании триглицеридов (главная часть жиров). Это риск развития ишемической болезни сердца (инфаркт миокарда) в первом десятилетии жизни и в возрасте 30-40 лет.
Комбинированная семейная гиперлипидемия - повышение уровней холестерина и триглицеридов. Во взрослом возрасте очень рано возникает атеросклероз коронарных и периферических сосудов.
Семейнаягиперлипидемия I типа характеризуется болями в животе, увеличением печени и селезенки, образованием желтых пятен на коже.
Семейнаягиперлипидемия II типа - наблюдается либо раннее развитие атеросклероза с тяжелым поражением коронарных сосудов, либо умеренное изменение липидов и развитие атеросклероза в возрасте 30 - 40 лет.
Семейнаягиперлипидемия III типа или гипертриглицеридемия наблюдается при диете с высоким содержанием углеводов. Рано выявляются желтые пятна на коже, атероматоз, выраженная коронарная болезнь.
Семейнаягиперлипидемия IV типа наблюдается также при повышенном содержании углеводов в диете. Клинически выявляются нарушения терпимости к глюкозе, ишемическая болезнь.
Семейнаягиперлипидемия V типа клинически характеризуется приступами болей в животе, увеличением печени и селезенки, развитием атеросклероза и ИБС, нарушением терпимости к глюкозе.
Значение болезни и ее форм определяется тем, что процесс усвоения липидов тесно связан с возникновением атеросклероза и ишемической болезни сердца.
65. синтез кетоновых тел. Роль кетоновых тел. Пути утилизации кетоновых тел в переферических тканях. Биосинтез холестерина и его производных. Роль холестерина в организме.
Под термином «кетоновые (ацетоновые) тела» подразумевают ацетоуксусную кислоту (ацетоацетат) СН3СОСН2СООН, β-оксимасляную кислоту (β-оксибутират, или D-3-гидроксибутират) СН3СНОНСН2СООН и ацетон СН3СОСН3. В здоровом организме ацетон в крови присутствует в крайне низких концентрациях, образуется в результате спонтанного декарбоксилированияацетоацетата и, по-видимому, не имеет определенного физиологического значения.
Кетоновые тела образуются в печени. Прежние представления о том, что кетоновые тела являются промежуточными продуктами β-окисления жирных кислот, оказались ошибочными.
Во-первых, в обычных условиях промежуточными продуктами β-окисления жирных кислот являются КоА-эфиры этих кислот, например β-оксибутирил-КоА, ацетоацетил-КоА.
Во-вторых, β-оксибутирил-КоА, образующийся в печени при β-окислении жирных кислот, имеет L конфигурацию, в то время как β-оксибутират, обнаруживаемый в крови, представляет собой D-изомер. Именно β-оксибутират D-конфигурации образуется в ходе метаболического пути синтеза β-окси-β-метилглутарил-КоА (3-гидрокси-3-метилглутарил-КоА).
На первом этапе из 2 молекул ацетил-КоА образуется ацетоацетил-КоА. Реакция катализируется ферментом ацетил-КоА-ацетилтрансферазой (3-кетотиолазой). Затем ацетоацетил-КоА взаимодействует еще с одной молекулой ацетил-КоА. Реакция протекает под влиянием фермента гидроксиметилглутарил-КоА-синтетазы. Образовавшийся β-окси-β-метилглутарил-КоА способен под действием гидроксиметилглутарил-КоА-лиазы
расщепляться на ацетоацетат и ацетил-КоА. Ацетоацетат восстанавливается при участии НАД-зависимой D-3-гидроксибутиратдегидрогеназы, при этом образуется D-β-оксимасляная кислота (D-3-гидроксибутират). Следует подчеркнуть, что фермент специфичен по отношению к D-стереоизомеру и не действует на КоА-эфиры. Существует второй путь синтеза кетоновых тел. Образовавшийся путем конденсации 2 молекул ацетил-КоАацетоацетил-КоА способен отщеплять коэнзим А и превращаться в ацетоацетат. Этот процесс катализируется ферментом ацетоацетил-КоА-гидролазой (деацилазой).
Однако второй путь образования ацетоуксусной кислоты (ацетоацетата) не имеет существенного значения, так как активность деацилазы в печени низкая.
В настоящее время ясна молекулярная основа изречения, что «жиры сгорают в пламени углеводов». Известно, что ацетил-КоА, образовавшийся при окислении жирных кислот, включается в цикл трикарбоновых кислот в условиях, когда расщепление жиров и углеводов соответствующим образом сбалансировано. Включение ацетил-КоА в цикл Кребса зависит от доступности оксалоацетата для образования цитрата. Однако если расщепление жиров преобладает, судьба ацетил-КоА изменяется. Объясняется это тем, что в отсутствие углеводов или при нарушении их использования концентрация оксалоацетата снижается. При голодании или диабете оксалоацетат расходуется на образование глюкозы и поэтому не может конденсироваться с ацетил-КоА. В таких условиях путь метаболизма ацетил-КоА отклоняется в сторону образования ацетоацетата и D-3-гидроксибутирата, т.е. кетоновых тел.
В крови здорового человека кетоновые тела содержатся лишь в очень небольших концентрациях (в сыворотке крови 0,03–0,2 ммоль/л).
При патологических состояниях (у лиц с тяжелой формой сахарного диабета, при голодании, а также у животных с экспериментальным острым стрептозотоциновым или аллоксановым диабетом) концентрация кетоновых тел в сыворотке крови увеличивается и может достигать 16–20 ммоль/л.
Следует подчеркнуть важную роль кетоновых тел в поддержании энергетического баланса. Кетоновые тела – поставщики «топлива» для мышц, почек и действую, возможно, как часть регуляторного механизма с обратной связью, предотвращая чрезвычайную мобилизацию жирных кислот из жировых депо. Печень в этом смысле является исключением, она не использует кетоновые тела в качестве энергетического материала. Как отмечалось, основным местом образования ацетоацетата и 3-гидроксибутирата служит печень. Из митохондрий печени эти соединения диффундируют в кровь и переносятся к периферическим тканям. Действительно, сердечная мышца и корковый слой почек предпочтительно используют в качестве «топлива» ацетоацетат, а не глюкозу.
Известно, что в периферических тканях 3-гидроксибутират (β-оксимасляная кислота) способен окисляться до ацетоацетата, а последний активируется с образованием соответствующего КоА-эфира (ацетоацетил-КоА).
Ацетоацетат может быть активирован путем переноса КоА с сукцинил-КоАв реакции, катализируемой специфической КоА-трансферазой. Образовавшийся ацетоацетил-КоА далее расщепляется тиолазой с образованием 2 молекул ацетил-КоА, которые затем включаются в цикл Кребса.
Биосинтез холестерина.
В синтезе холестерина можно выделить три основные стадии: I – превращение активного ацетата в мевалоновую кислоту, II – образование сквалена из мевалоновой кислоты, III – циклизация сквалена в холестерин.
Рассмотрим стадию превращения активного ацетата в мевалоновуюкислоту. Начальным этапом синтеза мевалоновой кислоты из ацетил-КоАявляется образование ацетоацетил-КоА посредством обратимой тиолазной реакции.
Затем при последующей конденсации ацетоацетил-КоА с 3-й молекулой ацетил-КоА при участии гидроксиметилглутарил-КоА-синтазы (ГМГ-КоАсинтаза) образуется β-гидрокси-β-метилглутарил-КоА.
Далее β-гидрокси-β-метилглутарил-КоА под действием регуляторного фермента НАДФ-зависимой гидроксиметилглутарил-КоА-редуктазы(ГМГ-КоА-редуктаза) в результате восстановления одной из карбоксильных групп и отщепления HS-KoA превращается в мевалоновую кислоту.
ГМГ-КоА-редуктазная реакция – первая практически необратимая реакция в цепи биосинтеза холестерина. Она протекает со значительной потерей свободной энергии (около 33,6 кДж). Установлено, что данная реакция лимитирует скорость биосинтеза холестерина.
На II стадии синтеза холестерина мевалоновая кислота превращается в сквален. Реакции II стадии начинаются с фосфорилированиямевалоновойкислоты с помощью АТФ. В результате образуется 5-фосфорный эфир, а затем 5-пирофосфорный эфир мевалоновой кислоты.
5-пирофосфомевалоновая кислота в результате последующего фосфорилирования третичной гидроксильной группы образует нестабильный промежуточный продукт – 3-фосфо-5 пирофосфомевалоновую кислоту, которая, декарбоксилируясьи теряя остаток фосфорной кислоты, превращается в изопентенилпирофосфат. Последний изомеризуется в диметилаллилпирофосфат.
Затем оба изомерных изопентенилпирофосфата (диметилаллилпирофосфат и изопентенилпирофосфат) конденсируются с высвобождением пирофосфата и образованием геранилпирофосфата.
К геранилпирофосфату вновь присоединяется изопентенилпирофосфат. В результате этой реакции образуется фарнезилпирофосфат.
В заключительной реакции данной стадии в результате НАДФН-зависимой восстановительной конденсации 2 молекул фарнезилпирофосфатаобразуется сквален.
На III стадии биосинтеза холестерина сквален под влиянием скваленоксидоциклазыциклизируется с образованием ланостерина. Дальнейший процесс превращения ланостерина в холестерин включает ряд реакций, сопровождающихся удалением трех метильных групп, насыщением двойной связи в боковой цепи и перемещением двойной связи в кольце В из положения 8, 9 в положение 5, 6 (детально эти последние реакции еще не изучены).
Значение для организма
Материнское молоко богато холестерином. Грудные и растущие дети нуждаются в богатых жирами и холестерином продуктах, в том числе для полноценного развития мозга и нервной системы. По данным радионуклидных исследований, в организме человека содержится до 350 граммов этого вещества: 90% в тканях и 10% -- в крови. Около 20% от общего количества холестерина в организме содержится в головном и спинном мозге -- он является структурным компонентом миелиновой «изоляции» нервов. В плазме крови он находится в виде сложных эфиров с высшими жирными кислотами и служит переносчиком при их транспорте.
В печени из холестерина синтезируются желчные кислоты, необходимые для эмульгирования и всасывания жиров в тонком кишечнике. На эти цели уходит 60-80% ежедневно образующегося в организме холестерина. Холестерин является «сырьем» для производства стероидных гормонов коры надпочечников (гидрокортизона и альдостерона), а также женских и мужских половых гормонов (эстрогенов и андрогенов). У мужчин помешательство на бесхолестериновых продуктах может быть опасным для сексуальной активности, а у женщин, слишком активных в борьбе с холестерином, нередко наступает аменорея. Особенно бессмысленно соблюдение безхолестериновой диеты женщинами детородного возраста, поскольку до наступления менопаузы женские половые гормоны просто не дают холестерину откладываться на стенках сосудов.
Холестерин -- важный компонент клеточной мембраны.
66. причины и типы гипо- и геперлипопротеинемий. Атеросклероз, этапы атерогенеза. Функции холестерина в организме человека. Основные направления терапии атеросклероза. Профилактика атеросклероза.
Согласно классификации ВОЗ, принято выделять пять типов гиперлипопротеинемий (ГЛП): I, IIа, IIб, III, IV, V, отличающихся нарушением обмена тех или иных ЛП. На практике врачу чаще приходится встречаться с типами IIа, IIб, IV. Классификация удобна тем, что описывает спектр ЛП при наиболее распространенных вариантах ГЛП. Однако здесь не принимаются во внимание первичные (генетически предопределенные) и вторичные (обусловленные как ответ на факторы окружающей среды или основное заболевание) причины нарушений. В этой классификации не учитывается концентрация холестерина ЛПВП, хотя его содержание существенно влияет на вероятность развития ишемической болезни сердца у больных с гиперлипидемией.
Необходимо помнить, что тип ГЛП у пациента может измениться с одного на другой под влиянием диеты, изменения массы тела и лечения.
Система фенотипирования, несмотря на недостатки, привлекла внимание к природе метаболических нарушений гиперлипидемий и позволила искать рациональные подходы к их диагностике и лечению.
ГИПЕРЛИПОПРОТЕИНЕМИИ (ГЛП)
I тип: гиперхиломикронемия
II тип: гипер-β-липопротеинемия
III тип: дис-β-липопротеинемия
IV тип: гипер-пре-β-липопротеинемия
V тип: гиперхиломикронемия и гипер-пре-β-липопротеинемия
ДРУГИЕ ТИПЫ ГЛП И ДЛП, НЕ ВОШЕДШИЕ В ОСНОВНУЮ КЛАССИФИКАЦИЮ
Гипер-α-липопротеинемия
Гипo-α-липопротеинемия
Ан-α-липопротеинемия
Гипо-β-липопротеинемия
Атеросклероз - прогрессирующее накопление избытка липопротеинов и других компонентов крови, сопровождающееся реактивным образованием фиброзной ткани преимущественно во внутренней оболочке артерий эластического и мышечно‑эластическоготипов, атакжекомплексомдругихизмененийвних.
В результате атеросклеротического поражения сужается просвет артерий, нарушается кровоснабжение органов и тканей, развиваются осложнения в виде кальциноза и аневризм стенок сосудов, тромбоза, эмболий и др.
Наиболее поражаемые атеросклерозом регионы сосудистого русла: брюшной отдел аорты, коронарные артерии, сонные артерии, артерии мозга, почечные артерии, артерии брыжейки и нижних конечностей.
Атеросклероз является разновидностью артериосклероза - его атероматозной формой.
Известно не менее 250 факторов, способных быть причиной и/или условиями, способствующими возникновению и развитию атеросклероза. В связи с трудностью чёткого разделения различных патогенных факторов на причины и условия их обозначают как факторы риска.
К наиболее значимым факторам риска относят курение, сахарный диабет, артериальную гипертензию, ожирение, гиперхолестеринемию (отношение ЛПНП к ЛПВП более 5:1), гипертриглицеридемию, гиподинамию, инсульты и заболевания ССС в семейном анамнезе, приём пероральных контрацептивов.
Выделяют следующие этапы атерогенеза: инициация его, прогрессирование атерогенеза, формирование атеромы, образование фиброатеромы, развитие осложнений атеросклероза.
1.Этап инициации атерогенеза заключается в повреждении и активации эндотелиальных клеток и экспрессии молекул адгезии на их поверхности. Этот этап носит неспецифический характер. Его признаки могут быть выявлены уже на 8–10–м году жизни.
Основные механизмы:
- Активация синтеза и выделения на поверхность эндотелиоцитов молекул адгезии (селектинов Р и Е, ICAM1, ICAM2, VCAM, интегринов и др.), а также кахексина. Это обусловливает: адгезию на поверхности эндотелиоцитовмононуклеарных клеток (в основном моноцитов), а также тромбоцитов и проникновение моноцитов и тромбоцитов в субэндотелиальное пространство.
- Транспорт в субэндотелиальное пространство ЛП крови. Там ЛП взаимодействуют с молекулами гликозаминогликанов. Находящиеся в интерстиции ЛП, особенно связанные с макромолекулами матрикса, подвергаются дальнейшим изменениям (окислению, ацетилированию, восстановлению, образованию перекисей, альдегидов и пр.). Особую роль в модификации ЛП играет СПОЛ. В аорте и других артериях человека обнаружены липооксигеназы - ферменты, катализирующие образование перекисей липидов. При распаде перекисей липидов накапливаются свободные радикалы, способные неэнзиматически инициировать СПОЛ. В стенке сосуда ЛП изолированы от антиоксидантных факторов плазмы крови (аскорбиновой кислоты, уратов, сульфгидрильных групп белков), поэтому особенно подвержены модификации в ходе СПОЛ. Окисляться может не только липидная, но и белковая часть ЛП (апоЛП), необходимая для взаимодействия с рецепторами ЛП. Модифицированные липиды служат хемоаттрактантами для лейкоцитов, а также подвергаются фагоцитозу макрофагами. Макрофаги с большим количеством ЛП в цитоплазме получили название пенистых клеток. Такое название связано с тем, что при обработке срезов ткани из макрофагов вымываются липиды. Под микроскопом вакуоли, образовавшиеся после удаления липидов, напоминают пену.
2.Этапы прогрессирования атерогенеза:
- Миграция в участки интимы артерий с повреждёнными и активированными эндотелиальными клетками большого числа моноцитов и тромбоцитов.
- Активация синтеза лейкоцитами, тромбоцитами, эндотелиоцитами БАВ (факторов хемотаксиса, кининов, Пг, факторов роста, факторов некроза опухолей), а также образования активных форм кислорода и липопероксидов. Указанные факторы потенцируют повреждение эндотелия и подэндотелиального слоя артерий.
Описанные выше механизмы и изменения интимы обозначаются как этап начального повреждения артерии, дисфункции эндотелия. Последующие изменения в стенке артерий относятся к этапу липидных пятен и полосок.
- Усиление поглощения макрофагами (мигрировавшими в интиму моноцитами) модифицированных ЛП при помощи так называемых «скевенджер‑рецепторов» (англ. scavengerreceptor). Скевенджер‑рецепторымакрофаговсвязываютсяпреимущественносмодифицированнымиЛПНП.
- Макрофаги, насыщенные липидами (в основном, эфирами холестерина), превращаются в пенистые клетки.
- Миграция из средней оболочки артерий в зону повреждения интимы ГМК, их пролиферация и синтез ими БАВ.
- Липидные полоски и пятна содержат пенистые клетки, лимфоциты (В‑и T-лимфоциты, хелперы, киллеры, пролиферирующиеГМК, тромбоциты, внеклеточныеЛП, коллаген, эластин, протеогликаны, гликозаминогликаны, продуцируемыеГМК.
3.Переходный этап атерогенеза - липосклеротический - характеризуется нарастанием процессов поступления ЛП винтиму, их модификации, образования и распада пенистых клеток. Это приводит к значительному увеличению содержания в интерстициальном пространстве модифицированных ЛП и компонентов соединительной ткани.
Все этапы атерогенеза, включая переходный, клинически могут ещё никак не проявляться.
4.Формирование атеромы и фиброатеромы обусловлено:
- Массированным проникновением моноцитов крови винтиму артерии.
- Увеличением масштаба миграции из средней оболочки сосуда ГМК, их пролиферации и приобретение ими синтетического фенотипа (трансформация).
- Прогрессирующей активацией синтеза компонентов межклеточного вещества соединительной ткани (протеогликанов, гликозаминогликанов, коллагеновых и эластических волокон).
Причины:
- Продолжающееся действие факторов риска.
- Формирование и/или активация по ходу атерогенеза факторов, потенцирующих повреждение стенки артерии (например, свободных радикалов, липопероксидов, ФНО, аутоагрессивныхIg и лимфоцитов).
Эффекты цитокинов: увеличение миграции моноцитов винтиму, стимуляция таксиса, пролиферации и трансформации ГМК, усиление захвата макрофагами ЛП, активация синтеза ГМК компонентов межклеточного вещества и началом формирования фиброзной крышки атеромы.
Атерома характеризуется:
- Наличием значительного количества клеточных элементов: пенистых клеток, ГМК на разных этапах пролиферации и трансформации, лимфоцитов, гранулоцитов, тромбоцитов.
- Массивными скоплениями внеклеточных ЛП - формированием ядра атеромы.
Фиброатерома - в дополнение к свойствам атеромы - характеризуется:
- Формированием фиброзной крышки над липидным ядром.
- Развитием сети микрососудов, окружающих атеросклеротический очаг.
Атеромы и особенно фиброатеромы выступают в просвет артерии, уменьшают его, а также стимулируют тромбообразование.
Этап развития осложнений атеросклероза:
5.Модификация атером и особенно фиброатером приводит к:
- Накоплению в них кальция и его соединений (особенно - в крышке фиброатеромы). Этот процесс получил название кальцификации, атерокальциноза.
- Трещинам крышки фиброатеромы и/или её изъязвлением, что сопровождается высвобождением содержимого атеромы в просвет артерии и развитием: пристеночного тромба (с угрозой обтурации артерии), эмболии.
- Разрыв стенок новообразованных микрососудов по периметру атеромы или фиброатеромы. Это может привести к: кровоизлияниям в стенку артерии, образованию пристеночных и интрамуральных тромбов.
6.Наиболее частые и значимые клинические осложнения атеросклероза:
- Инфаркты органов (сердца, мозга, почки, лёгких и других).
- Кровоизлияния и кровотечения.
- Ишемия органов и тканей (включая ИБС, ишемический инсульт, ишемию почек, стенки кишечника и конечностей). Ишемия органов развивается вследствие: сужения просвета артерии (атеромой, фиброатеромой, пристеночным тромбом, эмболом), сокращения ГМК артериол (под влиянием сосудосуживающих веществ, выделяемых клетками в области атеросклеротических изменений (лейкотриенов, эндотелина, тромбоксана А2, вазоконстрикторныхПг).
Профилактика и лечение.
Борьба с ожирением: вес всегда следует поддерживать в норме, так как только таким образом удастся предупредить развитие таких патологий как: ишемическая болезнь сердца, сахарный диабет и гипертония; Повышение физических нагрузок: дает возможность снизить общую массу тела, восстановить работу сердца и процесс кровообращения в данной области, предупреждает возникновение сахарного диабета и гипертонии; Правильно питание, снижение до минимума продуктов питания с большим количеством холестерина: специальная диета, состоящая из большого количества продуктов обогащенных ненасыщенными жирными кислотами и низким количеством животных жиров, переедание недопустимо; Отказ от спиртных напитков и курения: основные методы предупреждения развития данного заболевания. Как табаку, так и спиртным напиткам свойственно оказывать негативное воздействие не только на состояние сосудов, но еще и на функционирование сердца; Устранение переутомления и стрессовых ситуаций: дает возможность восстановить работу как эндокринной, так и нервной системы. Помимо этого восстанавливается нормальный обмен веществ.
В общем, чтобы предупредить развитие данной патологии либо вылечить ее необходимо четко следовать «определенному» режиму, то есть соблюдать все правила здорового образа жизни. Если больному удастся избавиться от одного фактора риска данной патологии, тогда он снизит риск развития ее осложнений в два раза. Избавление от всех факторов – это прямой путь к нормальному течению данного заболевания без возможных осложнений.
Ловастатин (мевакор, мефакор, новемакор) - препарат снижает общий холестерин, холестерин низкой плотности и триглицериды, повышает уровень холестерина высокой плотности. Показано, что ловастатин вызывает уменьшение атеросклеротических бляшек в сосудах сердца, правда, для этого требуется не менее 4-5 лет непрерывного применения.
Симвастатин (зокор) - уже через два недели снижает общий холестерин на 10%, а холестерин липопротеидов низкой плотности - на 33%, а также повышает холестерин высокой плотности на 12%. Он несколько активнее, чем ловастатин и лучше всасывается в кишечнике.
Правастатин (правахол, липревил, липостат) - по механизму действия похож на ловастатин, но биодоступность очень невелика (17-18%), хуже всасывается, чем другие статины, меньше влияет на холестерин высокой плотности. Период полувыведения (Т1/2) равен 1,5-3 часа. Побочные эффекты такие же как у других статинов.
Флувастатин (лескол) - первый синтетический препарат из группы статинов. Дает меньше побочных эффектов, чем другие статины. Т1/2=30 мин. Хорошо всасывается, но обильная пища уменьшает его биодоступность. Эффект достигается уже к концу первой недели, достигает максимума через 3-4 нед. и держится на этом уровне весь последующий период лечения. На сегодняшний день является одним из наиболее привлекательных препаратов.
67.основные пути обмена белков, переваривание белков и всасывание продуктов их распада, биологическая ценность белков.
белки определяют микро- и макроструктуру отдельных субклеточных образований, специфику организации клеток, органов и целостного организма (пластическая функция), в значительной степени динамическое состояние между организмом и окружающей его средой. Белковый обмен строго специфичен, направлен и настроен, обеспечивая непрерывность воспроизводства и обновления белков организма. В течение всей жизнедеятельности в организме постоянно и с высокой скоростью совершаются два противоположных процесса: распад, расщепление органических макромолекул и надмолекулярных структур и синтез этих соединений. Эти процессы обеспечивают катаболические реакции и создание сложной структурной
организации живого из хаоса веществ окружающей среды, причем ведущую роль в последнем случае играют именно белки. Все остальные виды обмена подчинены этой глобальной задаче живого – самовоспроизведению себе подобных путем программированного синтеза специфических белков. Для
осуществления этого используются энергия обмена углеводов и липидов, строительный материал в виде углеродных остатков аминокислот, промежуточных продуктов метаболизма углеводов и др.
Белки способны также выполнять энергетическую функцию, особенно при избыточном их поступлении с пищей или в экстремальных ситуациях, когда белки тела подвергаются усиленному распаду, восполняя недостаток питательных веществ, например при голодании или патологии (сахарный диабет). Как известно, при сгорании 1 г белков освобождается энергия, равная 16,8 кДж. Эта энергия обычно может быть полностью заменена энергией окисления углеводов и липидов, однако при длительном исключении последних из пищи у животных не наблюдается существенных патологических отклонений, тогда как исключение белков из пищи даже на короткий срок приводит к выраженным нарушениям, а иногда и к необратимым патологическим явлениям.Если человек находятся на малобелковой диете, то у них очень быстро развивается белковая недостаточность – патологическое состояСледовательно, белки являются незаменимыми для организма веществами, выполняющими прежде всего пластическую функцию, уникальную каталитическую функцию,принимают непосредственное участие в биосинтезе ряда гормонов и других биологически активных соединений, регулирующих процессы обмена веществ в организме.Следовательно, именно белковый обмен координирует, регулирует и интегрирует многообразие химических превращений в целостном живом организме, подчиняя его задачам сохранения вида и обеспечивая тем самым непрерывность жизни.
Характерной особенностью белкового обмена является его чрезвычайная разветвленность. Достаточно указать, что в обмене 20 аминокислот ,входящих в состав белковых молекул, в организме животных участвуют сотни промежуточных метаболитов, тесно связанных с обменом углеводов и липидов. Число ферментов, катализирующих химические реакции азотистого обмена, также исчисляется сотнями.
БИОЛОГИЧЕСКАЯ ЦЕННОСТЬ БЕЛКОВ
Состояние белкового обмена целостного организма зависит не только от количества принимаемого с пищей белка, но и от качественного состава его. Чем ближе аминокислотный состав принимаемого пищевого белка к аминокислотному составу белков тела, тем выше его биологическая ценность.
С понятием биологической ценности белков тесно связан вопрос об эссенциальных (незаменимых) аминокислотах. Живые организмы существенно различаются в зависимости от их способности синтезировать аминокислоты или другие азотсодержащие соединения, которые они могут использовать для биосинтеза аминокислот. Высшие позвоночные животные не синтезируют все необходимые аминокислоты. В организме человека и белых крыс синтезируются только 10 из 20 необходимых аминокислот – так называемые заменимые аминокислоты. Они могут быть синтезированы из продуктов обмена углеводов и липидов. Остальные 10 аминокислот не синтезируются в организме, поэтому они были названы жизненно необходимыми, эссенциальными, или незаменимыми аминокислотами.
Исключение какой-либо незаменимой аминокислоты из пищевой смеси сопровождается развитием отрицательного азотистого баланса, истощением, остановкой роста, нарушениями функции нервной системы и др.
Биологическая ценность пищевого белка целиком зависит от степени егоусвоения организмом, что в свою очередь определяется соответствием между аминокислотным составом потребляемого белка и аминокислотным составом белков организма. Такой пищевой белок лучше используется организмом для синтеза белков тканей. Для человека, например, белки мяса, молока, яиц биологически более ценны, поскольку их аминокислотный состав ближе к аминокислотному составу органов и тканей человека.
Однако это не исключает приема растительных белков, в которых содержится необходимый набор аминокислот, но в другом соотношении. Поэтому для обеспечения биосинтеза необходимого количества эндогенных белков человеку потребуется значительно больше растительных белков, чем животных.
Таким образом, для нормального роста и гармоничного развития организма человека исключительно большое значение имеют составление и подбор пищевых продуктов, содержащих оптимальный аминокислотный состав и обеспечивающих физиологически полноценное питание для разных
групп населения с учетом не только возраста и пола, но и различных климатических условий, характера труда, сезона года и т.д.
переваривание белков.
Главными источниками белков для человека являются пищевые продукты животного и растительного происхождения.Весь сложный процесс переваривания пищевых белков в пищеварительном тракте «настроен» таким образом, чтобы путем последовательного действия протеолитических ферментов лишить белки пищи видовой и тканевой специфичности и придать продуктам распада способность всасываться в кровь через стенку кишечника. Примерно 95–97% белков пищи всасывается в виде свободных аминокислот. Следовательно, ферментныйаппарат пищеварительного тракта осуществляет поэтапное, строго избирательное расщепление пептидных связей белковой молекулы вплоть до конечных продуктов гидролиза белков – свободных аминокислот. Гидролиз заключается в разрыве пептидных связей —СО—NH— белковой молекулы.Протеолитические ферменты (протеиназы) обладают широкой специфичностью действия, определяемой как размером полипептида, так и структурой радикалов аминокислот, участвующих в образовании пептидной связи.
Следует подчеркнуть, что с пищей человек получает огромное разнообразие белков, однако все они подвергаются воздействию ограниченного числа протеиназ. Эти ферменты относятся к классу гидролази часто называются также пептидазами. Известны две группы пептидаз: экзопептидазы, катализирующие разрыв концевой пептидной связи с освобождением одной какой-либо концевой аминокислоты, и эндопептидазы, преимущественно гидролизующие пептидные связи внутри полипептидной цепи.
В желудке имеются все условия для переваривания белков. Во-первых, в желудочном соке содержится активный фермент пепсин. Во-вторых, благодаря наличию в желудочном соке свободной соляной кислоты для действия пепсина создается оптимальная среда (рН 1,5–2,5). Следует особо указать на существенную роль соляной кислоты в переваривании белков: она переводит неактивный пепсиноген в активный пепсин, создает оптимальную среду для действия пепсина; в присутствии соляной кислоты происходят набухание белков, частичная денатурация и, возможно, гидролиз сложных белков. Кроме того, соляная кислота стимулирует выработку секретина в двенадцатиперстной кишке, ускоряет всасывание железа и
оказывает бактерицидное действие.Пепсин, катализирующий гидролиз пептидных связей, образованных
остатками ароматических аминокислот, расщепляет практически все природные белки. Исключение составляют некоторые кератины, протамины, гистоны и мукопротеины. При их гидролизе образуются различного размера пептиды и, возможно, небольшое число свободных аминокислот.
Дальнейшее превращение белков пищи осуществляется в тонкой кишке, где на белки действуют ферменты панкреатического и кишечного соков. Трипсин и химотрипсин действуют на белки аналогично пепсину, разрывают другие внутренние пептидные связи; оба фермента наиболее активны в слабощелочной среде (рН 7,2–7,8). Благодаря гидролитическому действию на белки всех трех эндопептидаз (пепсин, трипсин, химотрипсин) образуются различной длины пептиды и некоторое количество свободных
аминокислот. Дальнейший гидролиз пептидов до свободных аминокислот осуществляется под влиянием группы ферментов – пептидаз. Помимо панкреатическойкарбоксипептидазы, на пептиды действуют кишечная аминопептидаза и разнообразные дипептидазы. Эта группа ферментов относится к экзопептидазам.Точкой приложения аминопептидазы является пептидная связь с N-конца пептида. Карбоксипептидаза разрывает пептидную связь с противоположного С-конца пептида. Эти ферменты отщепляют по одной аминокислоте от полипептида.
В итоге остаются дипептиды, на которые действуют специфические дипептидазы, при этом образуются свободные аминокислоты, которые затем всасываются. Из других ферментов протеолиза следует упомянуть об эластазеи коллагеназе поджелудочной железы, гидролизующих соответственно эластин и коллаген. Топографически основные процессы гидролиза белков, как и углеводов и жиров, протекают на поверхности слизистой оболочки кишечника.
Всасывание.
Продукты гидролиза белков всасываются в пищеварительном тракте в основном в виде свободных аминокислот.Некоторые аминокислоты обладают способностью конкурентно тормозить всасывание других аминокислот, что свидетельствует о вероятном существовании общей переносящей системы или одного общего механизма. Так, в присутствии лизина тормозится всасывание аргинина, но не изменяется всасывание аланина, лейцина и глутамата. Существует два представления, по-видимому, дополняющих друг друга о том, что требуемая для активного транспорта энергия образуется за счет биохимических реакций (это так называемый направляемый метаболизмом транспорт) или за счет энергии переноса другого транспортируемого вещества, в частности энергии движения ионов Na+ (или других ионов) в клетку.
68. общие пути обмена аминокислот. Значение реакций дезаминирования, трансаминирования, декарбоксилирования. Диагностическоезнаачениетрансамилаз в сыворотке крови.
Общие пути обмена аминокислот
Общие пути превращения аминокислот включают реакции дезаминирования, трансаминирования, декарбоксилирования, биосинтеза и рацемизации.Реакции рацемизации характерны только для
микроорганизмов; открыты ферменты, катализирующие рацемизацию ряда аминокислот (Ала, Глу, Про, Мет, Лиз, Сер) и эпимеризациюоксипролинаи α,ε-диаминопимелиновой кислоты. Физиологическая роль рацемазмикроорганизмов сводится, вероятно, к синтезу D-изомеров аминокислот для построения клеточной оболочки.
Дезаминирование аминокислот
Доказано существование 4 типов дезаминирования аминокислот (отщепление аминогруппы). Выделены соответствующие ферментные системы, катализирующие эти реакции, и идентифицированы продукты реакции. Во всех случаях NH2-группа аминокислоты освобождается в виде аммиака.
Помимо аммиака, продуктами дезаминирования являются жирные кислоты, оксикислоты и кетокислоты. Для животных тканей, растений и большинства аэробных микроорганизмов преобладающим типом реакций является окислительноедезаминирование аминокислот, за исключением гистидина, подвергающегося внутримолекулярному дезаминированию. Рассмотрим более подробно механизм окислительногодезаминирования аминокислот, протекающего в две стадии.Первая стадия является ферментативной и завершается образованием
неустойчивого промежуточного продукта (иминокислота), который на второй стадии спонтанно без участия фермента, но в присутствии воды распадается на аммиак и α-кетокислоту. Следует указать, что оксидазы
аминокислот (L- и D-изомеров) являются сложными флавопротеинами, содержащими в качестве кофермента ФМН или ФАД, которые выполняют в этой реакции роль акцепторов двух электронов и протонов, отщепляющихся от аминокислоты. Оксидазы L-аминокислот могут содержать как ФМН, так и ФАД, а оксидазы D-аминокислот – только ФАД в качестве простетической группы.Восстановленныефлавиннуклеотиды оксидаз L- и D-аминокислот могут непосредственно окисляться молекулярным кислородом. При этом образуется перекись водорода, которая подвергается расщеплению под действием каталазы на воду и кислород.Помимо перечисленных 4 типов дезаминирования аминокислот и ферментов, катализирующих эти превращения, в животных тканях и печени человека открыты также три специфических фермента (серин- и треониндегидратазы и цистатионин-γ-лиаза), катализирующих неокислительное
дезаминирование соответственно серина, треонина и цистеина.Конечными продуктами реакции являются пируват и α-кетобутират, аммиак и сероводород. Поскольку указанные ферменты требуют присутствия пиридоксальфосфата в качестве кофермента, реакция неокислительного дезаминирования, вероятнее всего, протекает с образованием шиффовых оснований как промежуточных метаболитов.
Трансаминирование аминокислот
Под трансаминированием подразумевают реакции межмолекулярного переноса аминогруппы (NH2—) от аминокислоты на α-кетокислоту без промежуточного образования аммиака.Реакции трансаминирования являются обратимыми и, как выяснилосьпозже, универсальными для всех живых организмов. Эти реакции протекают при участии специфических ферментов, названных аминоферазами.Для реакций трансаминированияхарак-
терен общий механизм. Специфичность трансаминаз обеспечивается белковым компонентом. Ферменты трансаминирования катализируют перенос NH2-группы не на α-кетокислоту, а сначала на кофермент пиридоксальфосфат. Образовавшееся промежуточное соединение (шиффово основание) подвергается внутримолекулярным превращениям (лабилизацияα-водородного атома, перераспределение энергии связи), приводящим к освобождению α-кетокислоты и пиридоксаминфосфата; последний на второйстадии реакции реагирует с любой другой α-кетокислотой, что через те же стадии образования промежуточных соединений (идущих в обратном направлении) приводит к синтезу новой аминокислоты и освобождению
пиридоксальфосфата.
Декарбоксилирование аминокислот
Процесс отщепления карбоксильной группы аминокислот в виде СО2 получил название декарбоксилирования. Несмотря на ограниченный круг аминокислот и их производных, подвергающихся декарбоксилированию в животных тканях, образующиеся продукты реакции – биогенныеамины – оказывают сильное фармакологическое действие на множествофизиологических функций человека и животных. В животных тканях установлено декарбоксилирование следующих аминокислот и их производных: тирозина, триптофана, 5-окситриптофана, валина, серина, гистидина, глутаминовой и γ-оксиглутаминовой кислот, 3,4-диоксифенилаланина, цистеина, аргинина, орнитина, S-аденозилметионина и α-аминомалоновойкислоты. Помимо этого, у микроорганизмов и растений открыто декарбоксилирование ряда других аминокислот.
В живых организмах открыты 4 типа декарбоксилирования аминокислот:
1. α-Декарбоксилирование, характерное для тканей животных, при ко-
тором от аминокислот отщепляется карбоксильная группа, стоящая по
соседству с α-углеродным атомом. Продуктами реакции являются СО2
и биогенные амины.
2. ω-Декарбоксилирование, свойственное микроорганизмам. Например,
из аспарагиновой кислоты этим путем образуется α-аланин.
3. Декарбоксилирование, связанное с реакцией трансаминирования.
В этой реакции образуются альдегид и новая аминокислота, соот-
ветствующая исходной кетокислоте.
4. Декарбоксилирование, связанное с реакцией конденсации двух мо-
Лекул.
Эта реакция в тканях животных осуществляется при синтезе δ-аминолевулиновой кислоты из глицина и сукцинил-КоАи при синтезе сфинголипидов, а также у растений при синтезе биотина.
Реакции декарбоксилирования в отличие от других процессов промежуточного обмена аминокислот являются необратимыми. Они катализируются специфическими ферментами – декарбоксилазами аминокислот, отличающимися от декарбоксилазα-кетокислот (см. главу 10) как белковым компонентом, так и природой кофермента. Декарбоксилазы аминокислот состоят из белковой части, обеспечивающей специфичность действия, и простетической группы, представленной пиридоксальфосфатом (ПФ), как и у трансаминаз.
диагностическое значение трансаминаз.
Диагностическая тактика при незначительном повышении уровня трансаминаз у больных без каких-либо клинических проявлений является актуальной и распространенной проблемой, с которой ежедневно сталкивается большое количество практикующих врачей. Интерпретация результатов исследования аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ) в сыворотке крови как обязательного скринингового рутинного теста.Чаще всего врачу общей практики приходится рассматривать и интерпретировать повышение уровня АСТ и/или АЛТ у пациентов с ожирением, сахарным диабетом, дислипопротеидемиями либо с полным отсутствием симптомов какого-либо заболевания. В то же время у пациентов с нормальным уровнем АЛТ и АСТ могут наблюдаться тяжелые поражения печени, сопровождающиеся хроническим повреждением гепатоцитов (обширный некроз, цирроз, гепатит С), за счет значительного уменьшения количества клеток, синтезирующих трансаминазы.
АЛТ — нормальный уровень в сыворотке крови составляет 0,1–0,68 ммоль/ч на 1 л — является ферментом, катализирующим перенос аминогруппы аланина на α-кетоглутаровую кислоту с образованием пировиноградной и глутаминовой кислот. Переаминирование происходит в присутствие кофермента — пиридоксальфосфата — производного витамина В6. Наиболее высокая активность АЛТ выявляется в клетках печени, меньшая — в почках, поджелудочной железе, сердце и скелетных мышцах. АЛТ — это внутриклеточный фермент, его содержание в сыворотке крови здоровых людей невелико. Активность зависит от многих демографических факторов, в частности — пола (у женщин несколько ниже, чем у мужчин), массы тела (более высокие значения при ожирении) и расы (у европейцев АЛТ на 15% ниже, чем у азиатов и лиц негроидной расы).
За счет преимущественного расположения АЛТ в цитозоле печени его повышение считают более специфичным для заболеваний печени (Kundrotas L.W., Clement D.J., 1993). Но при повреждении или разрушении клеток, богатых АЛТ, происходит выброс этого фермента в кровяное русло, что приводит к повышению активности его в крови. При вирусных гепатитах степень увеличения активности АЛТ, как правило, пропорциональна тяжести заболевания. В острых случаях активность фермента в сыворотке крови может превышать нормальные значения в 5–10 раз и более. При вирусном гепатите повышение его активности происходит в очень ранние сроки — еще до появления желтухи. В динамике при благоприятном течении процесса активность АЛТ медленно снижается до исходных значений в течение нескольких недель. Быстрое снижение активности фермента в сочетании с нарастающей гипербилирубинемией свидетельствует о неблагоприятном прогнозе — переходе в хроническую форму, развитии фиброза, цирроза.
АСТ — нормальный уровень в сыворотке крови составляет 0,1–0,45 ммоль/ч на 1 л — также является внутриклеточным ферментом, который обратимо катализирует трансаминирование, межмолекулярный перенос аминогруппы с 1-аспарагиновой кислоты на α‑кетоглютаровуюкислоту. ACT содержитсявскелетныхмышцах, сердце, практическивовсехпаренхиматозныхорганах—печени, почках, головном мозге, поджелудочной железе, легких, в клетках крови — эритроцитах и лейкоцитах. Необходимо помнить, что результаты определения аспартатаминотрансферазы могут искажаться в сторону повышения — если в сыворотку попадает ACT из разрушенных эритроцитов, или в сторону понижения — когда в результате хранения в течение нескольких дней происходит инактивация фермента.
В гепатоцитах большая часть ACT (80% активности) обнаруживается в митохондриях и составляет митохондриальнуюизоформу, и лишь около 20% содержится в цитозольной фракции — цитозольнаяизоформа. Незначительные и умеренные воспалительные процессы в гепатоцитах сопровождаются высвобождением ACT из цитозоля, митохондриальные структуры повреждаются мало, поэтому общее количество попадающего в кровь ACT невелико, в отличие от уровня АЛТ, которая целиком локализуется в цитозоле и переходит в кровь при повреждении последнего.
При тяжелом поражении печени, массивных некрозах гепатоцитов ACT может высвобождаться из поврежденных митохондрий этих клеток, при этом показатели сывороточной ACT приобретают особую диагностическую и прогностическую ценность. Как следствие, в клинической практике помимо прямых значений АСТ и АЛТ зачастую используется соотношение АСТ/АЛТ, получившее название коэффициента Де Ритиса. В норме его значение составляет 1,3±0,4. Клиническая значимость соотношения АСТ/АЛТ в сыворотке крови неоднократно изучалась при различных заболеваниях печени. Результаты анализа современных публикаций показывают, что при неосложненных вирусных гепатитах или неалкогольных поражениях печени соотношение АСТ/АЛТ, как правило, не превышает 1 или снижается до 0,6–0,8. Напротив, повышение коэффициента более 1,4 (за счет повышения активности АСТ) наблюдается при циррозах, тяжелых алкогольных и токсических поражениях печени с разрушением большей части печеночных клеток либо при других заболеваниях (например, при болезни Вильсона коэффициент АСТ/АЛТ может превышать 4).
Лактатдегидрогеназа (ЛДГ) — нормальный уровень в сыворотке крови 140–280 Ед/л — представляет собой фермент, катализирующий обратимое восстановление пировиноградной кислоты до молочной в процессе гликолиза. Наибольшая активность ЛДГ обнаруживается в почках, миокарде, скелетных мышцах, печени. В организме ЛДГ представлена пятью органоспецифическими изоферментами, при этом последние являются различными комбинациями двух типов (H и М) полипептидных цепей. Органная специфичность изоферментов позволяет использовать их определение в сыворотке крови с целью топической диагностики.
69.катаболизм аминокислот возможные пути расщепления углеродного скелета, утилизация аминного азота, радикалов.
Трансаминирование и дезаминирование аминокислот ведет к образованию безазотистых углеродных скелетов аминокислот – α-кетокислот. В состав белков входят 20 аминокислот, различающихся по строению углеводородного радикала, каждый из которых катаболизируется по своим специфическим метаболическим путям.
Катаболизм всех аминокислот сводится к образованию шести веществ, вступающих в общий путь катаболизма: пируват, ацетил-КоА, a-кетоглутарат, сукцинил-КоА, фумарат, оксалоацетат.
Аминокислоты, которые превращаются в промежуточные продукты ЦТК (a-кетоглутарат, сукцинил-КоА, фумарат), и образуют в конечном итоге оксалоацетат, могут использоваться в процессе глюконеогенеза. Такие аминокислоты называются гликогенными. К ним относятся: аланин, аргинин, аспартат, глутамат, глицин, гистидин, метионин, пролин, серин, треонин, валин, цистеин.
Катаболизм лейцина и лизина не включает стадии образования пировиноградной кислоты, их углеводородная часть превращается непосредственно в ацетоацетат (лейцин, лизин) или в ацетил-КоА (лейцин) и используются в синтезе кетоновых тел.
Тирозин, фенилаланин, изолейцин и триптофан являются смешанными или одновременно гликогенными и кетогенными. Часть углеродных атомов их молекул при катаболизме образует пируват, другая часть включается в ацетил-КоА, минуя стадию пирувата.
Истинной кетогенной аминокислотой является лейцин.См №68.
70.биосинтез аминокислот: общие пути индивидуальные различия.
В организме человека возможен синтез восьми заменимых аминокислот: Ала, Асп, Асн, Сер, Гли, Глу, Глн, Про. Углеродный скелет этих аминокислот образуется из глюкозы. α-Аминогруппа вводится в соответствующие α-кетокислоты в результате реакций трансаминирования. Универсальным донором α-аминогруппы служит глутамат.
Путём трансаминированияα-кетокислот, образующихся из глюкозы, синтезируются аминокислоты.
Глутамат также образуется при восстановительном аминированииα-кетоглутаратаглутаматдегидрогеназой.
Эти реакции обратимы и играют большую роль как в процессе синтеза аминокислот, так и при их катаболизме. Такие реакции, выполняющие двойную функцию, называют амфиболическими.
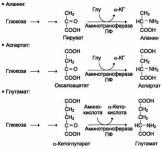
Амиды глутамин и аспарагин синтезируются из соответствующих дикарбоновых аминокислот Глу и Асп (см. схему А).
Серин образуется из 3-фосфоглицерата - промежуточного продукта гликолиза, который окисляется до 3-фосфопирувата и затем трансаминируется с образованием се-рина (см. схему Б).
Существует 2 пути синтеза глицина:
из серина с участием производного фолиевой кислоты в результате действия се-риноксиметилтрансферазы:

в результате действия фермента глицинсинтазы в реакции:

Пролин синтезируется из глутамата в цепи обратимых реакций. Эти же реакции используются и при катаболизме пролита.
Кроме восьми перечисленных заменимых аминокислот, в организме человека могут синтезироваться ещё четыре аминокислоты.
Частично заменимые аминокислоты Apr и Гис синтезируются сложным путём в небольших количествах. Большая их часть должна поступать с пищей.
Синтез аргинина происходит в реакциях орнитинового цикла.
Гистидин синтезируется из АТФ и рибозы. Часть имидазольного цикла гистидина - N=CH-NH- образуется из пуринового ядра аденина, источником которого служит АТФ, остальная часть молекулы - из атомов рибозы. При этом образуется 5-фосфорибозиламин, который кроме синтеза гистидина необходим для синтеза пуринов.

Для синтеза условно заменимых аминокислот тирозина и цистеина требуются незаменимые аминокислоты фенилаланин и метионин соответственно.
Образование других аминокислот также возможно при наличии соответствующих α-кетокислот,которые могут трансаминироваться с глутаматом. Таким образом, незаменимой частью молекулы аминокислот является их углеродный скелет. Источником таких незаменимых ос-кетокислот служат только белки пищи. Исключение составляют лизин и треонин, которые не подвергаются трансаминированию, их а-кетоаналоги с пищей практически не поступают и в организме не синтезируются. Единственный источник этих аминокислот - пищевые белки.
Схема А.
Схема Б.

Схема В.
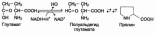
71.Квашиоркор - вид тяжёлой дистрофии. Болезнь обычно возникает у детей 1-4 лет, хотя бывает что она возникает и в более старшем возрасте (например у взрослых или у более старших детей). Педиатр с Ямайки Сисели Уильямс (англ. Cicely D.Williams) впервые описала это состояние в медицинском журнале Lancet в 1935 году. Когда ребенка кормят грудью, он получает определенные аминокислоты, необходимые для роста из материнского молока. Когда ребенка отлучают от груди, в случае, когда продукты, заменяющие материнское молоко, содержат много крахмалов и сахаров и мало белков (как это обычно случается в странах, где основная диета людей состоит из крахмалосодержащих овощей, или там, где начался массовый голод), у ребенка может начаться квашиоркор.
Один из симптомов - вздутость животов детей (асцит), часто возникающая у детей бедных районов Африки, объясняется тем, что клубни маниокa содержат лишь небольшое количество белка (1.2 %) и очень немногие незаменимые аминокислоты. При питании, основанном на маниоке, эти факторы приводят к детской пеллагре (квашиоркор). Из-за недостатка важных аминокислот внутренние органы накапливают воду. В связи с этим рекомендуется употребление также и листьев маниоки, содержащих большое количество белка, в качестве овоща.
Недостаточность белка, часто сочетающаяся с дефицитом энергии, витаминов и микроэлементов, приводит к нарушению развития ребенка, дистрофическим изменениям органов и тканей. Ведущим в патогенезе квашиоркора является недостаток в организме ребенка пластических веществ и ферментов, Особенно легко квашиоркор развивается у детей раннего возраста, т.к. для обеспечения интенсивных процессов роста и развития необходимо поддержание постоянного положительного азотистого баланса в организме (поступление белков должно превышать их расходование).
применение аминокислот в качестве лекарственных препаратов
Набор аминокислот с высоким процентом ß-аланина (коммерческое название «Аминовил - Р») зарегистрирован в России и во многих странах Европы, используется для устранения стресса и отека мозга, улучшения мозгового кровообращения. Композит снижает токсические проявления противоэпилептических препаратов и усиливает их терапевтический эффект. Следует отметить, что для снижения уровня глутаминовой кислоты в мозге на практике эффективны и другие аминокислоты, в частности лейцин. Механизм действия заключается в активации фермента глутаматдегидрогеназы, дезаминирующегоглутамат до α-кетоглутаровой кислоты - одного из компонентов цикла трикарбоновых кислот.
Важная роль в деятельности мозга принадлежит и аминокислоте глицину. Помимо участия в образовании важнейших биологически активных соединений - пуриновых нуклеотидов, гема, креатина и др., аминокислота выполняет роль тормозного нейромедиатора, контролируя процессы формирования тонкой моторики пластических процессов и тонусных реакций поперечно-полосатой мускулатуры. Основная масса глицина сосредоточена в спинном мозге, где аминокислота, высвобождаясь из окончаний клеток Реншоу, опосредует постсинаптическое высвобождение (торможение) мотонейронов. Поэтому препарат широко используется в неврологической практике для устранения повышенного мышечного тонуса.
Регуляция активности НМДА-глутаматных рецепторов также осуществляется глицином. Аминокислота имеет собственный сайт в составе большинства глутаматных возбуждающих рецепторов.
При взаимодействии с магнием глицин оказывает тормозящее воздействие, в свободном виде - стимулирующее воздействие.
Успешное использование глицина в качестве лекарственного препарата для лечения больных с острым нарушением мозгового кровообращения привлекло внимание неврологов к изучению механизма действия препарата.
Менее изучена роль диаминокарбоновой кислоты лизина как нейромедиатора. Предполагается, что точкой приложения медиатора является специфический рецептор в зоне каудального отдела ретикулярной формации нервной системы. Видимо поэтому при приеме внутрь даже в небольших дозах (0,5 г в сутки) лизина быстро устраняется спазм поперечно-полосатой мускулатуры.
Таким образом, из представленных данных видно что с помощью аминокислотных композитов можно коррегировать содержание возбуждающих и тормозящих нейромедиаторов в клетках мозга. На практике это означает необходимость использования природных метаболитов для эффективного устранения не только функциональных заболеваний нервной системы (стресс, неврозы), но и тяжелых поражений (острое нарушение мозгового кровообращения, дегенеративные заболевания).
Классификация используемых композитов.
1.Композиты для коррекции пирамидной недостаточности(Глюкаприм-Р, Примавит-Р, Дехол). Содержат набор аминокислот в различных соотношениях: глицин, треонин, g-аминомасляная кислота; метаболиты – цитрат натрия, сукцинат натрия, oксолат натрия.
2. Композиты для коррекции экстрапирамидной недостаточности (Витамикст-Р, Аминокомпозит-Р, Олдарин, Аминопуринол). Содержат набор аминокислот в различных соотношениях: глутамин, аспарагиновая кислота, лейцин, изолейцин, цистин и цистеин и др.
3. Композиты комбинированного действия (Нейродин, Нейровит-Р, Глюканал-F). Содержат набор аминокислот в различных соотношениях: лизин, аргинин, фенилалаламин, тирозин, орнитин и др.
4. Другие композиты (Аминовил-Р, Провит-Р, Цереброн, Севит). Содержат набор аминокислот в различных соотношениях: α-аланин, глицин, глутамин, аспарагановая кислота ассоциированная с магнием, сквален и др.
72.
73.Обмен фенилаланина и тирозина
Фенилаланин относится к незаменимым аминокислотам, поскольку ткани животных не обладают способностью синтезировать его бензольное кольцо. В то же время тирозин полностью заменим при достаточном поступлении фенилаланина с пищей. Объясняется это тем, что основной путь
превращения фенилаланина начинается с его окисления (точнее, гидроксилирования) в тирозин. Реакция гидроксилирования катализируется специфической фенилаланин-4-монооксигеназой, которая в качестве кофермента содержит, как все другие гидроксилазы, тетрагидробиоптерин. Блокирование этой реакции, наблюдаемое при нарушении синтеза фенилаланин-4-монооксигеназы в печени, приводит к развитию тяжелой наследственной болезни – фенилкетонурии (фенилпировиноградная
олигофрения). В процессе трансаминирования тирозин превращается в n-оксифенилпировиноградную кислоту, которая под действием специфической оксидазы подвергается окислению, декарбоксилированию, гидроксилированию и внутримолекулярному перемещению боковой цепи с образованием гомогентизиновой кислоты; эта реакция требует присутствия аскорбиновой кислоты, роль которой пока не выяснена. Дальнейшее превращение гомогентизиновой кислоты в малеилацетоуксусную кислоту
катализируется оксидазой гомогентизиновой кислоты. Малеилацетоуксусная кислота под действием специфической изомеразы в присутствии глутатиона превращается в фумарилацетоуксусную кислоту, подвергающуюся гидролизу с образованием фумаровой и ацетоуксусной кислот, дальнейшие превращения которых уже известны.Фенилаланин и тирозин являются также предшественниками меланинов. В этом важном биологическом процессе, обеспечивающем пигментацию кожи, глаз, волос, активное участие принимает фермент тирозиназа.
Фенилкетонурия
Фенилкетонурия, фенилпировиноградная олигофрения, наследственное заболевание из группы ферментопатий, в основе которого лежит аномалия аминокислотного обмена вследствие отсутствия или резкого снижения активности фермента фенилаланингидроксилазы. Описана в 1934 норв. учёным А. Фёллингом (A.Foiling) (болезнь Фёллинга). Частота Ф. – 1 случай на 10–15 тыс. новорождённых; наследуется по аутосомно-рецессивному типу. При Ф. фенилаланингидроксилаза сохраняет только около 5% активности, в связи с чем нарушается обмен фенилаланина и вследствие этого – тирозина, триптофана и др., накапливаются промежуточные продукты обмена – фенилэтиламин, фенилпировиноградная кислота и др. и возникает дефицит метаболитов, необходимых для нормального функционирования организма В частности, вероятная причина умственных расстройств – дефицит медиаторов нервной системы (адреналина, норадреналина, серотонина и др.). Т. о., при Ф. возникает комплекс взаимосвязанных метаболических расстройств, состоящий из первичного ферментного нарушения и обусловленных им др. нарушений обмена.
Ф. проявляется главным образом выраженной олигофренией (идиотией или имбецильностью). Диагностируется в первые дни жизни ребёнка с помощью экспресс-методов – микробиологических или биохимических. Последние основаны на определении пировиноградной кислоты в моче посредством индикаторов (проба Фёллинга). Лечение сводится главным образом к специальной диете (резкое ограничение продуктов, содержащих фенилаланин).
74.пути обезвреживания аммиака в организме. Цикл мочевины.
В организме человека подвергается распаду около 70 г аминокислот в сутки, при этом в результате реакций дезаминирования и окисления биогенных аминов освобождается большое количество аммиака, являющегося высокотоксичным соединением. Поэтому концентрация аммиака в организме должна сохраняться на низком уровне. Действительно, уровень аммиака в крови в норме не превышает 60 мкмоль/л.Аммиак должен подвергаться связыванию в тканях с образованием нетоксичных соединений, легко выделяющихся с мочой.Один из путей связывания и обезвреживания аммиака в организме,
в частности в мозге, сетчатке, почках, печени и мышцах,– это биосинтез глутамина (и, возможно, аспарагина). Глутамин и аспарагин выделяются с мочой в небольшом количестве. Было высказано предположение, что они выполняют скорее транспортную функцию переноса аммиака в нетоксичной форме.Энергетически синтез аспарагина обходится организму дороже, поскольку образовавшийся РРi далее распадается на ортофосфат. Часть аммиака легко связывается сα-кетоглутаровой кислотой благодаря обратимости глутаматдегидрогеназной реакции. Если учесть связывание одной молекулы аммиака при синтезе глутамина, то нетрудно видеть, что в организме имеется хорошо функционирующая система,
связывающая две молекулы аммиака. Глутамин, кроме того, используется почками в качестве резервного
источника аммиака (образуется из глутамина под действием глутаминазы), необходимого для нейтрализации кислых продуктов обмена при ацидозе и защищающего тем самым организм от потери с мочой используемых для этих целей ионов Na+.
Орнитиновый цикл мочевинообразования
Основным механизмом обезвреживания аммиака в организме является биосинтез мочевины. Последняя выводится с мочой в качестве главного конечного продукта белкового, соответственно аминокислотного, обмена. На долю мочевины приходится до 80–85% от всего азота мочи. Основным и, возможно, единственным местом синтеза мочевины является печень. Весь цикл мочевинообразования может быть представлен следующим образом. На первом этапе синтезируется макроэргическое соединение карбамоилфосфат – метаболически активная форма аммиака, используемая в качестве исходного продукта для синтеза пиримидиновых нуклеотидов (соответственно ДНК и РНК) и аргинина (соответственно белка и мочевины).Реакция требует затраты двух молекул АТФ, открыта в митохондриях
клеток печени и используется преимущественно для синтеза аргинина
и мочевины. В этой реакции в качестве активного стимулирующего аллостерического эффектора действует N-ацетилглутамат. Вторую, также необратимую, реакцию катализирует глутаминзависимаякарбамоилфосфатсинтетаза. На втором этапе цикла мочевинообразования происходит конденсация
карбамоилфосфата и орнитина с образованием цитруллина; реакцию катализирует орнитикарбамоилтрансфераза.На следующей стадии цитруллин превращается в аргинин в результате
двух последовательно протекающих реакций. Первая из них, энергозависимая,– это конденсация цитруллинаи аспарагиновой кислоты с образованием аргининосукцината (эту реакцию катализирует аргининосукцинатсинтетаза). Аргининосукцинат распадается в следующей реакции на аргинин и фумарат при участии другого фермента – аргининосукцинатлиазы.
На последнем этапе аргинин расщепляется на мочевину и орнитин под действием аргиназы.
Суммарная реакция синтеза мочевины без учета всех промежуточных
продуктов может быть представлена в следующем виде:
СO2 + NH3 + ЗАТФ + 2Н2O + Аспартат –> Мочевина + 2АДФ + АМФ + Фумарат + 2Рi + РРi.
Данная реакция сопровождается снижением свободной энергии (ΔG0 = –40 кДж), поэтому процесс всегда протекает в направлении синтеза мочевины. Следует указать, что синтез мочевины энергетически дорого
обходится организму. На синтез одной молекулы мочевины требуетсязатрата четырех высокоэнергетических фосфатных групп: две молекулы АТФ расходуются на синтез карбамоилфосфата и одна – на образование аргининоянтарной кислоты, при этом АТФ расщепляется на АМФ и РРi, который при гидролизе также образует две молекулы Рi.
Учитывая известные фактические данные о механизмах обезвреживания аммиака в организме, можно сделать следующее заключение. Часть аммиака используется на биосинтез аминокислот путем восстановительного аминированияα-кетокислот по механизму реакции трансаминирования. Аммиак связывается при биосинтезе глутамина и аспарагина. Некоторое количество аммиака выводится с мочой в виде аммонийных солей. В форме креатинина, который образуется из креатина и креатинфосфата, выделяется из организма значительная часть азота аминокислот. Наибольшее количество аммиака расходуется на синтез мочевины, которая выводитсяс мочой в качестве главного конечного продукта белкового обмена в организме человека и животных. Подсчитано, что в состоянии азотистого равновесия организм взрослого здорового человека потребляет и соответственно выделяет примерно 15 г азота в сутки; из экскретируемогос мочой количества азота на долю мочевины приходится около 85%, креатинина – около 5%, аммонийных солей – 3%, мочевой кислоты – 1% и на другие формы – около 6%.
75.Направление и интенсивность обмена белков в первую очередь определяются физиологическим состоянием организма и несомненно регулируются, как и все другие виды обмена, нейрогормональными факторами. Более интенсивно обмен белков протекает в детском возрасте, при активной мышечной работе, беременности и лактации, т.е. в случаях, когда резко повышаются потребности в белках. Существенное влияние на белковый обмен оказывает характер питания и, в частности, количественный и качественный белковый состав пищи. При недостаточном поступлении белков с пищей происходит распад собственных белков ряда тканей (печени, плазмы крови, слизистой оболочки кишечника и др.) с образованием свободных аминокислот, обеспечивающих синтез абсолютно необходимых цитоплазматических белков, ферментов, гормонов и других биологически активных соединений. Таким образом, «в жертву» приносятся некоторые «строительные» белки тканей для обеспечения жизнедеятельности целостного организма. Введение с пищей повышенных количеств белка, напротив, не оказывает заметного влияния на состояние белкового обмена, поскольку избыток белка не откладывается про запас, а в виде конечных продуктов азотистого обмена выводится с мочой. Более существенное значение имеет, однако, качественный белковый состав пищи, так как отсутствие или недостаток хотя бы одной какой-либо незаменимой аминокислоты может служить лимитирующим фактором биосинтеза всех белков в организме.
Синтез белка подчиняется закону «все или ничего» и осуществляется при условии наличия в клетке полного набора всех 20 аминокислот. Даже при поступлении всех аминокислот с пищей организм может испытывать состояние белковой недостаточности, если всасывание какой-либо одной аминокислоты в кишечнике замедлено или если она разрушается в большей степени, чем в норме, под действием кишечной микрофлоры. В этих случаях будет происходить ограниченный синтез белка или организм будет компенсировать недостаток аминокислоты для биосинтеза белка за счет распада собственных белков. Степень усвоения белков и аминокислот пищи зависит также от количественного и качественного состава углеводов и липидов, которые резко сокращают энергетические потребности организма за счет белков. Экспериментальный и клинический материал свидетельствует, что диета с недостаточным содержанием жиров и низкокалорийная пища способствуют повышению экскреции аминокислот и продуктов их распада с мочой.
Имеются экспериментальные доказательства прямой и опосредованной связи белкового обмена с обеспеченностью организма витаминами, в частности В1, В2, В6, РР и др. Обмен белков регулируется, кроме того, деятельностью желез внутренней секреции. Гормоны определяют в известной мере направление (в сторону синтеза или распада) и интенсивность белкового обмена. Например, после введения АКТГ и гормонов щитовидной железы наблюдается интенсивный распад тканевых белков. Другие
Таким образом, состояние белкового обмена определяется множеством факторов, как экзогенных (окружающая среда, характер питания и др.), так и эндогенных (физиологическое состояние организма, включающее нервно-гормональный статус, ферментная оснащенность и др.). Любые отклонения от нормального физиологического состояния организма отражаются на азотистом обмене. Знание закономерностей изменений обмена белков при данном конкретном патологическом процессе – необходимая предпосылка для правильного выбора тактики терапевтических мероприятий по устранению нарушенного процесса обмена.
Азотистый баланс
Баланс азота в организме (разность между количеством потребляемого и выделяемого азота) — один из широко используемых индикаторов белкового обмена. У здорового человека скорости анаболизма и катаболизма находятся в равновесии, поэтому азотистый баланс равен нулю. При травме или при стрессе, например при ожогах, потребление азота снижается, а потери азота повышаются, вследствие чего у больного азотистый баланс становится отрицательным. При выздоровлении азотистый баланс должен становиться положительным вследствие получения белка с пищей. Исследование азотистого баланса даёт более полную информацию о состоянии пациента, имеющего метаболические потребности в азоте. Оценка экскреции азота у критических больных позволяет судить о количестве азота, потерянного в результате протеолиза.
Для оценки азотистого баланса используют два способа измерения потерь азота с мочой:
■измерениеазотамочевинывсуточноймочеирасчётныйметод определения общей потери азота;
■прямоеизмерениеобщегоазотавсуточноймоче.
Общий азот включает все продукты обмена белков, выводимые с мочой. Количество общего азота сопоставимо с азотом усвоенного белка и составляет примерно 85% азота, поступившего с белками пищи. Белки содержат в среднем 16% азота, следовательно, 1 г выделенного азота соответствует 6,25 г белка. Определение суточного выделения азота мочевины с мочой позволяет удовлетворительно оценивать величину азотистого баланса (АБ) при максимально возможном учёте поступления белка: АБ = [поступивший белок (г)/6,25] - [суточные потери азота мочевины (г) + 3], где число 3 отражает приблизительные потери азота с калом и др.
Этот показатель (АБ) является одним из самых надёжных критериев оценки белкового обмена организма. Он позволяет своевременно выявить катаболическую стадию патологического процесса, оценить эффективность коррекции питания и динамику анаболических процессов. Установлено, что в случаях коррекции выраженного катаболического процесса необходимо довести АБ с помощью искусственного питания до +4-6 г/сут. Важно следить за экскрецией азота изо дня в день
Прямое определение общего азота в моче предпочтительнее исследования азота мочевины, особенно у критических больных. Выделение общего азота с мочой в норме составляет 10-15 г/сут, его процентное содержание распределяется следующим образом: 85% — азот мочевины, 3% — аммония, 5% — креатинина, 1% — мочевой кислоты. Расчёт АБ по общему азоту проводят по следующей формуле: АБ = [поступивший белок (г)/6,25] --[суточные потери общего азота (г) + 4].
Определение общего азота в моче во время начальной катаболической стадии необходимо проводить через день, а затем 1 раз в неделю.
Важный критерий, дополняющий все приведённые выше, — определение экскреции креатинина и мочевины с мочой.
Экскреция креатинина отражает метаболизм мышечного белка. Нормальная экскреция креатинина с суточной мочой составляет 23 мг/кг для мужчин и 18 мг/кг для женщин. При истощении мышечной массы наблюдается снижение экскреции креатинина с мочой и уменьшение кре-атинин-ростового индекса. Гиперметаболический ответ, имеющий место у большинства больных с неотложными состояниями, характеризуется возрастанием общих метаболических расходов, что ускоряет потерю мышечной массы. У таких пациентов в состоянии катаболизма главная задача поддерживающего питания заключается в сведении к минимуму потерь мышечной массы.
Экскреция мочевины с мочой широко используют для оценки эффективности парентерального питания с использованием источников аминного азота. Уменьшение выделения мочевины с мочой следует считать показателем стабилизации трофического статуса.
Результаты лабораторных тестов позволяют определить группы риска по развитию осложнений, вызванных недостаточностью питания и воспалительными реакциями, у больных, находящихся в критическом состоянии, в частности, с помощью расчёта прогностического воспалительного и питательного индекса (PrognosticInflamatoryandNutritionalIndex — PINI) по следующей формуле [Ingenbleek Y., Carpenter Y.A., 1985]: PINI = [Кислый a1-гликопротеин (мг/л)хСРБ (мг/л)]/[альбумин (г/л)хпреальбумин (мг/л)]. В соответствии с индексом PINI группы риска распределяются следующим образом:
■ниже 1 —здоровоесостояние;
■ 1-10 —группанизкогориска;
■ 11-20 —группавысокогориска;
■более 30 —критическоесостояние.
76.Нуклеиновые кислоты составляют существенную небелковую часть сложного класса органических веществ, получивших название нуклеопротеиновпоследние являются основой наследственного аппарата клетки
хромосом. Белковые компоненты нуклеопротеинов подвергаются многообразным превращениям, аналогичным метаболизму белков и продуктов их распада – аминокислот. Помимо уникальной роли нуклеиновых кислот в хранении и реализации наследственной информации, промежуточные продукты их обмена, в частности моно-, ди- и трифосфатнуклеозиды, выполняют важные регуляторные
функции, контролируя биоэнергетику клетки и скорость метаболических процессов. В то же время нуклеиновые кислоты не являются незаменимыми пищевыми факторами и не играют существенной роли в качестве энергетического материала.
Переваривание нуклеопротеинов и всасывание продуктов их распада осуществляются в пищеварительном тракте. Под влиянием ферментов желудка, частично соляной кислоты, нуклеопротеины пищи распадаются на полипептиды и нуклеиновые кислоты; первые в кишечнике подвергаются гидролитическому расщеплению до свободных аминокислот. Распад нуклеиновых кислот происходит в тонкой кишке в основном гидролитическим путем под действием ДНК- и РНКазы панкреатического сока.
Продуктами реакции при действии РНКазы являются пуриновые и пиримидиновые мононуклеотиды, смесь ди-и тринуклеотидов и резистентные к действию РНКазыолигонуклеотиды. В результате действия ДНКазы
образуются в основном динуклеотиды, олигонуклеотиды и небольшое количество мононуклеотидов. Полный гидролиз нуклеиновых кислот до стадии мононуклеотидов осуществляется, очевидно, другими, менее изученными ферментами (фосфодиэстеразами) слизистой оболочки кишечника.
В отношении дальнейшей судьбы мононуклеотидов существует два предположения. Считают, что мононуклеотиды в кишечнике под действием неспецифических фосфатаз (кислой и щелочной), которые гидролизируютфосфоэфирную связь мононуклеотида («нуклеотидазное» действие), расщепляются с образованием нуклеозидов и фосфорной кислоты и в таком виде всасываются. Согласно второму предположению, мононуклеотиды всасываются, а распад их происходит в клетках слизистой оболочки
кишечника. Имеются также доказательства существования в стенке кишечника нуклеотидаз, катализирующих гидролитический распад мононуклеотидов. Дальнейший распад образовавшихся нуклеозидов осуществляется внутри клеток слизистой оболочки преимущественно фосфороли-
тическим, а не гидролитическим путем.
Всасываются преимущественно нуклеозиды, и в таком виде частьазотистых оснований может быть использована для синтеза нуклеиновыхкислот организма. Если происходит дальнейший распад нуклеозидов до свободных пуриновых и пиримидиновых оснований, то гуанин не используется для синтетических целей.
Таким образом, синтез нуклеиновых кислот, мономерными единицами которых являются мононуклеотиды, будет определяться скоростью синтеза пуриновых и пиримидиновых нуклеотидов; синтез последних в свою очередь зависит от наличия всех составляющих из трех компонентов. Источником рибозы и дезоксирибозы служат продукты превращения глюкозы в пентозофосфатном цикле. Пока не получены доказательства существенной роли пищевых пентоз в синтезе нуклеиновых кислот. Фосфорная кислота также не является лимитирующим фактором, поскольку она поступает в достаточном количестве с пищей. Следовательно, биосинтез нуклеиновых кислот начинается с синтеза азотистых оснований (точнее,
мономерных молекул – мононуклеотидов).
77.Биосинтез пуриновых нуклеотидов
Пуриновые основания, образующиеся в процессе переваривания нуклеиновых кислот в кишечнике, в дальнейшем практически не используются, поэтому их синтез осуществляется из низкомолекулярных предшественников, продуктов обмена углеводов и белков. Впервые работами Дж. Бьюкенена, Дж. Гринберга экспериментально доказано включение ряда меченых атомов, в частности 15N- и 14С-глицина, 15N-аспартата, 15N-глутамина и др., в пуриновое кольцо мочевой кислоты.
Синтез инозиновой кислоты начинается с D-рибозо-5-фосфата, который, как известно, является продуктом
пентозофосфатного цикла и на которыйпереносится в необычной реакции пирофосфатная группа АТФ. Образовавшийся 5-фосфорибозил-1-пирофосфат (ФРПФ) взаимодействует сглутамином, являющимся донором NH2-группы, в результате чего образуется β-5-фосфорибозил-амин, причем в процессе реакции наряду с освобождением пирофосфата и свободной глутаминовой кислоты происходит изменение его конфигурации (из α- в β-). Таким образом, данная стадия становится ключевой реакцией в синтезе
пуринов. На следующей стадии присоединяется вся молекула глицинак свободной NH2-группе β-5-фосфорибозил-амина (реакция нуждается в доставке энергии АТФ) с образованием глицинамидрибонуклеотида. Затем, на следующей стадии, цепь удлиняется за счет присоединения формильнойгруппы из N5,N10-метенил-ТГФК с образованиемформилглицинамидрибонуклеотида. На формильную группу последнего переносится далее амидная группа глутамина и синтезируется формилглицинамидинрибонуклеотид (реакция также идет с потреблением энергии АТФ). На следующей стадии замыкается пятичленное имидазольное кольцо и образуется 5-аминоимидазолрибонуклеотид, который способен акцептировать СО2с образованием рибонуклеотида 5-аминоимидазол-4-карбоновой кислоты.
В последующем двухступенчатом процессе, в котором участвуют аспарагиновая кислота и АТФ, образуется 5-аминоимидазол-4-карбоксамидрибонуклеотид и освобождается фумаровая кислота. В этих реакциях азот
аспарагиновой кислоты включается в 1-е положение будущего пуринового ядра. Последний углеродный атом пиримидинового остатка кольца пурина вводится в виде формильного остатка (источник N10-формил-ТГФК), который присоединяется к 5-NH2-группе. После этого отщепляется молекула воды и второе кольцо замыкается. В результате образуется первый пуриновый нуклеотид – инозиновая кислота (ИМФ), которая является предшественником пуриновых нуклеотидов в составе нуклеиновых кислот.
АМФ и ГМФ образуются из ИМФ, причем в синтезе обоих мононуклеотидов участвуют по два фермента, различных по своему механизмудействия. Образование ГМФ из ИМФ катализируют ИМФ-дегидрогеназа
и ГМФ-синтетаза, а образование АМФ из того же предшественника катализируется последовательным действием аденилосукцинатсинтетазыи аденилосукцинатлиазы.
Регуляция биосинтеза пуринов
На синтез молекулы IMP затрачивается энергия гидролиза шести макроэргических фосфодиэфирных связей АТР, при этом в качестве предшественников выступают глицин, глутамин, метенилтетрагидрофолат и аспартат. Для экономии энергетических и питательных ресурсов важна эффективная регуляция процесса биосинтеза пуринов denovo. Важнейшую роль в этом процессе играет внутриклеточная концентрация ФРПФ. Она определяется соотношением скоростей его синтеза, утилизации и деградации. Скорость синтеза ФРПФ зависит от 1) наличия субстратов синтеза, особенно рибозо-5-фосфата, и 2) каталитической активности ФРПФ-синтазы, которая в свою очередь связана с внутриклеточной концентрацией фосфатов, а также с концентрацией пуриновых и пиримидиновых рибонуклеотидов, выступающих в роли аллостерических регуляторов. Скорость утилизации ФРПФ в значительной степени зависит от интенсивности цикла реутилизации пуриновых оснований, в ходе которого ксантин и гуанин фосфорибозилируютсядо соответствующих рибонуклеотидов. В меньшей степени скорость утилизации ФРПФ зависит от интенсивности синтеза пуринов denovo. Этот вывод основан на следующем наблюдении: в эритроцитах и культивируемых фибробластах мужчин с наследственным нарушением активности гипоксантин-гуанин—фосфо-рибозилтрансферазы уровень ФРПФ повышается в несколько раз.
Подагра или артрит подагрический - хроническое заболевание, возникающее в результате нарушения обмена веществ, отложения мочекислых солей (уратов) в тканях суставов, в костях, хрящах, сухожилиях.
Небезынтересно отметить, что подагра - одно из древнейших заболеваний, описанное впервые еще Гиппократом.
Подагрой страдали лучшие умы человечества – философы древности Ахилл и Эдип, Александр Македонский, Генрих VI, Кромвель, кардинал Мазарини, Иван Грозный, Петр I, Микеланджело, Пушкин, Тургенев, Мопассан, Стендаль, Колумб, Ньютон, Чарльз Дарвин. Генетики даже ввели такое понятие – гении подагрического типа.
Болезнь развивается почти исключительно у мужчин среднего возраста. Обычно наблюдаются артриты суставов нижних конечностей с частым вовлечением 1 пальца стопы, голеностопных и коленных суставов. Реже наблюдается артрит мелких суставов кистей и локтевых суставов. Подагрический артрит имеет характерные особенности: он часто развивается ночью, интенсивность боли нарастает очень быстро и за несколько часов достигает максимума. Боль обычно очень сильная, движения в суставе становятся невозможными, наблюдаются покраснение кожи и повышение температуры тканей над суставом. Может повышаться температура тела. Самостоятельно или под влиянием лечения артрит стихает за несколько дней, не оставляя в большинстве случаев никаких остаточных изменений. К факторам, провоцирующим возникновение приступа подагрического артрита, относят: чрезмерное употребление в пишу продуктов, богатых пуриновыми основаниями, главным образом мяса, алкоголя, операции, травмы, прием мочегонных средств. У 15—20% больных подагрой возникает мочекаменная болезнь (приступ почечной колики иногда может быть первым признаком подагры).Преобладание в пище животных жиров, ожирение, малоподвижный образ жизни, курение приводят к комплексным нарушениям обмена веществ. И как следствие – гипертония, сахарный диабет, остеоартроз, остеопороз, нарушение обмена холестерина и другие недуги. Подагра – в их числе.
Больным подагрой не следует пользоваться аспирином или другими салицилатами. Имейте в виду, что безрецептурные препараты, содержащие аспирин, могут привести к задержке в организме мочевой кислоты и обострению подагрического артрита. Аспирин также снижает эффективность противоподагрических препаратов.
Больные подагрой, впервые выявленной или в периоде обострения заболевания, подлежат стационарному лечению в специализированных ревматологических отделениях областных или городских больниц. Больные подагрой в период ремиссии заболевания при условии назначения адекватной терапии могут находиться под надзором ревматолога, нефролога по месту жительства в районных поликлиниках. Ориентировочная продолжительность лечения в стационарных условиях (специализированные ревматологические отделения) — 7-14 суток при условии подбора адекватной эффективной терапии, улучшение клинических и лабораторных признаков заболевания.
Лечение при подагре предусматривает:
по возможности быстрое и осторожное купирование острого приступа;
профилактику рецидива острого подагрического артрита;
профилактику или регресс осложнений болезни, вызванной отложением кристаллов однозамещенного урата натрия в суставах, почках и других тканях;
профилактику или регресс сопутствующих симптомов, таких как ожирение, гипертриглицеридемия или гипертензия;
профилактику образования мочекислых почечных камней.
Традиционные рекомендации по диете заключаются в ограничении потребления пуринов и алкоголя. К продуктам с высоким содержанием пуринов относятся мясные и рыбные продукты, а также чай, какао и кофе. Недавно также было показано, что снижение веса, достигаемое умеренным ограничением углеводов и калоража пищи в сочетании с пропорциональным повышением белка и ненасыщенных жирных кислот, приводило у больных подагрой к значительному уменьшению уровня мочевой кислоты и дислипидемии.
78.Биосинтез пиримидиновых нуклеотидов
Механизм синтеза пиримидиновых нуклеотидов почти полностью расшифрован благодаря исследованиям П. Рейхарда. Показано, что в клетках животных и в микроорганизмах конечными продуктами синтеза также не являются свободные пиримидиновые основания и остаток рибозы присоединяется к уже сформировавшемуся пиримидиновому кольцу. Синтез начинается с элементарных уровней (СО2, NH3, аспартат), и специфическую ключевую роль выполняет оротовая кислота.
Последовательность химических реакций синтеза пиримидиновых нуклеотидов, в частности УМФ, можно представить в следующем виде:

Оротидин-5'-фосфат (ОМФ)Уридин-5'-фосфат (УМФ)
Как видно, I стадия синтеза УМФ включает катализируемое цитоплазматическойкарбамоилфосфатсинтетазой образование карбамоилфосфата из глутамина.
На II стадии карбамоилфосфат реагирует с аспартатом, в результате чего образуется N-карбамоиласпарагиновая кислота. Последняя подвергается циклизации (под действием дигидрооротазы) с отщеплением молекулы воды, при этом образуется дигидрооротовая кислота, которая, подвергаясь дегидрированию, превращается в оротовую кислоту. В этой реакции участвует специфический НАД-содержащий фермент дигидрооротатдегидрогеназа. Оротовая кислота обратимо реагирует с ФРПФ,
являющимся донатором рибозо-фосфата, с образованием оротидин-5'-фосфата (ОМФ). Декарбоксилирование последнего приводит к образованию первого пиримидинового нуклеотида – уридин-5-фосфата (УМФ). Превращение УМФ в УДФ и УТФ осуществляется, как и пуриновых нуклеотидов, путем фосфотрансферазных реакций:
УМФ + АТФ <=> УДФ + АДФ;
УДФ + АТФ <=> УТФ + АДФ.
Регуляция синтеза пиримидиновых НУКЛЕОТИДОВ
Регуляторным ферментом в синтезе пиримидиновых нуклеотидов является полифункциональный
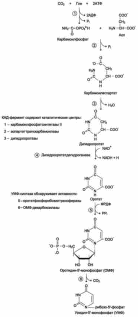
КАД-фермент. УМФ и пуриновые нуклеотиды аллостерически ингибируют, а ФРДФ активирует его карбамоилсинтетазную активность, тогда как активность аспартаттранскарбамоилазного домена ингибирует ЦТФ, но активирует АТФ.
Этот способ регуляции позволяет предотвратить избыточный синтез не только УМФ, но и всех других пиримидиновых нуклеотидов и обеспечить сбалансированное образование всех четырёх основных пуриновых и пиримидиновых нуклеотидов, необходимых для синтеза РНК.
79.БИОСИНТЕЗ ДЕЗОКСИРИБОНУКЛЕОТИДОВ
Синтез дезоксирибонуклеотидов идёт с заметной скоростью только в тех клетках, которые вступают в S-фазу клеточного цикла и готовятся к синтезу ДНК и делению. В покоящихся клетках дезоксинуклеотиды практически отсутствуют. Все дезоксинуклеотиды, кроме тимидиловых, образуются из рибонуклеотидов путём прямого восстановления ОН-группы у второго углеродного атома рибозы в составе рибонуклеозиддифосфатов до дезоксирибозы. Тимидиловыенук-леотиды синтезируются из dУМФ особым путём с участием N5,N10-метилен-Н4-фолата.
А. Рибонуклеотидредуктазный комплекс
Реакцию восстановления НДФ в дезоксипроизводные катализирует рибонуклеотидредук-тазный комплекс, в состав которого входят: собственно рибонуклеотидредуктаза (РНР), белок тиоредоксин и фермент тиоредоксинредуктаза, обеспечивающий регенерацию восстановленной формы тиоредоксина.
Рибонуклеотидредуктаза - олигомерный белок, состоящий из двух В1- и двух В2-субъединиц, и содержит негеминовое железо в качестве кофактора.
Непосредственным донором водорода в реакции восстановления рибозы служит низкомолекулярный белок тиоредоксин. В рабочую часть этого белка входят 2 SH-группы, которые, отдавая водород, окисляются с образованием дисульфидного мостика. Второй фермент комплекса - тиоредоксинредуктаза - катализирует гидрирование окисленного тиоредоксина с использованием NADPH.
При участии комплекса РНР образуются: dАДФ, dГДФ, dУДФ и dЦДФ, которые с помощью НДФ-киназ превращаются в дНТФ, 3 из которых (кроме дУДФ) непосредственно используются в синтезе ДНК.
дНДФ + АТФ → дНТФ + АДФ.
Б. Биосинтез тимидиловых нуклеотидов.
Тимидин-5'-монофосфат (дТМФ) образуется из дУМФ в реакции, катализируемой тимиди-латсинтазой (рис. 10-18). Донором метильной группы, появляющейся в 5-положении пиримидинового кольца в молекуле дТМФ, служит кофермент тимидилатсинтазы - N5,N10-метилен-Н4-фолат. С помощью этого кофермента в молекулу дУМФ включается метиленовая группа и восстанавливается в метальную, используя 2 атома водорода от Н4-фолата.
Образование субстрата тимидилатсинтазной реакции - дУМФ осуществляется двумя путями;
дефосфорилированиемдУДФ;
гидролитическим дезаминированиемдЦМФ с помощью дЦМФдезаминазы. дЦМФ получается при дефосфорилированиидЦДФ - одного из продуктов рибонуклеотидредуктаз-ной реакции. В организме человека это основной путь образования дУМФ.
Скорость синтеза дТМФ зависит также от количества второго субстрата тимидилатсинтазной реакции - N5,N10-метилен-Н4-фолата, пополнение запасов которого осуществляется при участии 2 ферментов: дигидрофолатредуктазы, которая с участием NADPH восстанавливает Н2-фолат в Н4-фолат, и серии гидроксиметилтрансферазы, осуществляющей перенос β-гидроксиметиленовой группы серина на Н4-фолат. У человека дТМФ образуется, главным образом, из дЦДФ.
"Запасные" пути синтеза дезоксирибонуклеотидов.
В быстроделящихся клетках наряду с синтезом дезоксинуклеотидов с помощью рибонуклеотид-редуктазного комплекса и тимидилатсинтазы активируются реакции, обеспечивающие повторное использование тимина и дезоксицитидина в реакциях, катализируемых ферментами "запасных" путей и обратимых реакций катаболизма. Под влиянием тимидинфосфорилазы протекает следующая реакция:
Тимин + Дезоксирибоза-1-фосфат → Тимидин + Н3Р04.
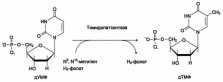

Тимидинкиназа катализирует следующую реакцию:
Тимидин + АТФ → дТМФ + АДФ.
Дезоксицитидинкиназа катализирует следующую реакцию:
Дезоксицитидин + АТФ → дЦМФ + АДФ.
80.ОБМЕН НУКЛЕИНОВЫХ КИСЛОТ
В клетках тканей нуклеиновые кислоты распадаются под влиянием ферментов. Ферменты, расщепляющие полинуклеотидные цепи, называют нуклеазами, или фосфодиэстеразами, так как они ускоряют реакции разрыва межнуклеотидныхфосфодиэфирных связей в молекулах нуклеиновых кислот. Различают эндонуклеазыиэкзонуклеазы.
Эндонуклеазы действуют на внутренние межнуклеотидные связи в молекулах ДНК и РНК. Таким образом, при их участии осуществляется деполимеризация нуклеиновых кислот, в основном до олигонуклеотидов.
Экзонуклеазы отщепляют нуклеотиды с 3′- или 5'-конца полинуклеотидной цепи и обеспечивают распад нуклеиновых кислот до свободных нуклеотидов.
По специфичности действия различают дезоксирибонуклеазы (ДНКазы,расщепляющие ДНК) и рибонуклеазы (РНКазы, гидролизующие РНК). В результате их действия образуются олигонуклеотиды и лишь небольшое количества мононуклеотидов. Рибо- и дезоксирибонуклеозидфосфаты расщепляются до нуклеозидов и фосфорной кислоты под действием фосфатаз (нуклеотидаз).
Нуклеозиды могут расщепляться гидролитическим путем с участием фермента нуклеозидазы:
Аденозин + Н2О → Аденин + рибоза
Пентозы окисляются до CO2 и H2O; фосфорная кислота используется для фосфорилирования органических соединений или выводится из организма. Азотистые основания превращаются в конечные продукты обмена и выделяются с мочой.
Продукты гидролиза нуклеиновых кислот поступают в клетки организма, где используются для синтеза нуклеотидов, нуклеиновых кислот, или же для удовлетворения энергетических потребностей клетки и организма.


