

3. Соотношения линейности Бренстеда-Поляни в катализе
Перспективный подход к предвидению каталитического действия твердых катализаторов основан на использовании соотношения линейности Бренстеда-Поляни, суть и основное значение которого состоит в том, что оно устанавливает связь между кинетическими (Е, k, ) и термодинамическими ( ) величинами, характерными для данного каталитического процесса.
Разные формы этого соотношения имеют вид:
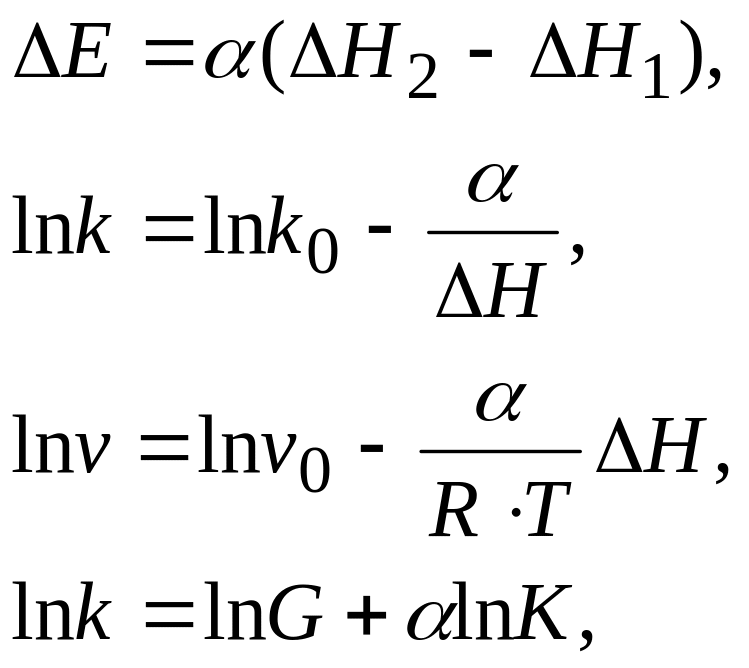 (5.12)
(5.12)
где , lnk0, lnv0 – постоянные эмпирические коэффициенты в ряду сходных катализаторов для данной реакции и в ряду однотипных реакций для данного катализатора; k, v – константа скорости и скорость реакции, К –константа равновесия, G – энергия Гиббса.
Тепловой эффект реакции () и энергия Гиббса (G) отражают величину энергии разрыва связи катализатор-субстрат.
Рассмотрим для примера реакцию окисления на оксидах металлов в качестве катализатора. Соотношение Бренстеда-Поляни используется в форме:
Е = Е0 +
![]() ,
(5.13)
,
(5.13)
где Е0 и – постоянные, одинаковые в ряду разных оксидов для заданной реакции окисления. Энергия активации реакции для разных катализаторов (Е) и теплота адсорбции кислорода на катализаторе ( ) (равная энергии связи кислорода (Q) с металлом).
Строится график в координатах Е – , определяется и Е0, зная которые можно рассчитать Е по известному или наоборот для катализаторов, с которыми не проводились опыты. По этим величинам можно судить об активности катализаторов. Корреляция между каталитической активностью и энергией связи кислорода справедлива не только для простых оксидов, но и для более сложных соединений типа шпинелей, солей ванадатов, молибдатов, смешанных оксидов и т.д.
Вопросы для самоконтроля
Что является мерой каталитической активности в гомогенном и ферментативном катализе? Каков порядок этой величины в каждом из типов катализа?
Какие величины используют для оценки эффективности катализатора в гетерогенном катализе?
В чем различие терминов «интегральная» и «дифференциальная» селективность? Ответ поясните.
Во сколько раз увеличиться скорость химической реакции при изменении активационного барьера на: а) 4 кДж/моль;б) 40 кДж/моль.
5.4 Механизмы гомогенных каталитических реакций
Промежуточное химическое взаимодействие реагентов с катализатором может протекать по двум видам механизма – слитно и раздельно.
При слитном механизме с состав активированного комплекса (промежуточного соединения) входят, наряду с катализатором, все реагирующие вещества и реакция идёт по схеме:
1. A + B
AB![]()
D + E без
катализатора,
D + E без
катализатора,
2. A + B +K ABK D + E + K c катализатором.
Увеличение скорости реакции в этом случае объясняется уменьшением энергии активации процесса
При раздельном механизме каталитический процесс осуществляется поэтапно:
3.
![]() .
.
4.
![]() .
.
Все соединения со значком ( ) означают активированный комплекс системы, находящейся в переходном состоянии. В этих реакциях суммарная энергия активации реакции необязательно должна уменьшаться, а скорость процесса растёт за счёт роста энтропийного фактора, так как при этом растёт количество стадий и упрощается строение активированных комплексов. Реакции с раздельным и слитным механизмом протекают при разных температурах; первые – при 600 – 800 К, а вторые – в области 300 - 400 К.
Как правило, катализатор снижает энергию активации реакции и ускоряет её. Замедлить реакцию (не цепную) катализатор не может, так как если в присутствии катализатора энергия активации растёт, то реакция пойдёт по обычному, не каталитическому пути и скорость её окажется прежней. Замедляющее действие катализатор может оказывать на цепную реакцию, когда он ускоряет стадию обрыва цепи.
Рассмотрим реакцию, протекающую по раздельному механизму [см. уравнения (3), (4)]. Согласно теории активированного комплекса, скорость реакции определяется скоростью распада комплекса на конечные продукты:
![]() . (5.14)
. (5.14)
В соответствии с принципом стационарности:
![]() . (5.15)
. (5.15)
Откуда:
![]() , (6.16)
, (6.16)
![]() , (5.17)
, (5.17)
![]() . (5.18)
. (5.18)
Подставляя (5.18) в (5.16) и затем в (5.14), находим:
 . (5.19)
. (5.19)
Это уравнение лежит в основе кинетики гетерогенно–каталитических реакций.
Из (5.19) видно, что скорость образования продукта реакции прямо пропорциональна концентрации катализатора сК, что хорошо согласуется с опытом.
Характерные частные случаи:
1) если k2k3,
т.е. константа скорости распада
промежуточного соединения АК на исходные
реагенты значительно превышает константу
скорости образования активированного
комплекса AKB![]() ,
то
,
то
![]() , (5.20)
, (5.20)
т.е. скорость реакции прямо пропорциональна концентрации катализатора и реагентов. Промежуточное вещество (АК) называется промежуточным веществом Аррениуса.
2) если k2 k3 , тогда всё количество промежуточного вещества превращается в конечные продукты и скорость реакции пропорциональна концентрации вещества (A), из которого образуется вещество (АК):
![]() . (5.21)
. (5.21)
Промежуточное вещество в этом случае носит название промежуточное вещество Вант Гоффа. Например, реакция окисления тиосульфата перекисью водорода в кислой среде с I– ионами
2S2O32- + H2O2 + 2H+ S4O62- + 2H2O
идёт по стадиям:
I– + H2O2 IO- + H2O,
I-+ IO– + 2H+ I2 + H2O,
I2 + 2S2O32– S4O62– + 2I–.
В этом случае IO– – промежуточное вещество Вант Гоффа. Если же в качестве катализатора взять молибденовую кислоту, то конечным продуктом её будут сульфат ионы и вода:
MoO4 + 4H2O2 [MoO8]2– + 4H2O,
[MoO8]2– + S2O32– +H2O 2SO42– + 2H+ + [MoO4]2–,
причём [MoO8]2– – является промежуточным веществом Аррениуса.