

Синдром Луї-Бар.
Синдром Луї-Бар характеризується наступними групами симптомів:
- телеангіектазіями шкіри та слиззових;
- змінами нервово-психічної сфери (мозочковий, стріо-паллідарний синдром та ін.);
- змінами з боку внутрішніх органів (хронічні запальні захворювання у зв'язку з вродженим дефіцитом імуноглобулінів);
- підвищеним рівнем хромосомних аберацій;
- високою частотою пухлинних уражень.
Епідеміологія, генетика
Ген синдрому Луї-Бар картований на хромосомі 11q 22-23. Тип успадкування аутосомно-рецесивний. Частота захворювання 1:40000.
Клініка та діагностика.
Телеангіектазії локалізуються на бульбарній кон’юнктиві, шкірі щік, у міжлопаточній ділянці. З'являються у віці від 2 до 6 років (рис.14,15). Також у хворих відзначаються прогеричні зміни шкіри та волосся. Навіть у маленьких дітей виявляють сиве волосся. Діти рано втрачають підшкірний "дитячий жир". Надалі з'являються ділянки атрофії, що нагадують сліди від вітряної віспи.
Серед неврологічних симптомів домінують атактичний (мозочковий), паллідарний та стріо-паллідарний. Симптоми ураження нервової системи з'являються рано, на 1-2 році життя. З віком відзначається їхнє клінічне наростання. Мозочкова атаксія завжди є першою й показовою ознакою захворювання. Вона починається в періоді дитинства та стає очевидною, коли дитина починає ходити. На більш пізніх стадіях у дітей з'являються міоклонічні посмикування кінцівок і тулуба, хореоатетоз. Обличчя набуває характерного виразу, з'являється гіпомімія. Поступово мозочковий симптомокомплекс змінюється паллідарним і стріопаллідарним синдромами, з'являється гіпокінезія, скутість.
У хворих із синдромом Луї-Бар виявляють порушення як у клітинній, так і гуморальній ланках імунітету, наслідком чого є хронічні бронхо-легеневі захворювання, отити, гайморити, пієлонефрити. Найбільш часто у хворих знижена концентрація IgA, IgG2 і/або IgE.
При комп'ютерній томографії і ЯМРТ виявляють атрофію мозочка.
Синдром Луї-Бар відноситься до синдромів хромосомної нестабільності. Цитогенетичною ознакою захворювання є висока частота невипадкових розривів і перебудов хромосом. Підвищений рівень хромосомних аберацій є важливим діагностичним критерієм захворювання. При цитогенетичному дослідженні виявляють підвищений рівень хромосомних аберацій у вигляді одиночних фрагментів, пробілів, парних фрагментів.
Серед неоплазій у хворих із синдромом Луї - Бар переважають злоякісні лімфоми, у тому числі лімфосаркоми, ретикулосаркоми, гістіосаркоми, лімфогрануломатоз. Частота злоякісних неоплазій лімфоретикулярної системи серед хворих із синдромом Луї - Бар становить 10-15%. Часто спостерігаються пухлини іншої локалізації. Біля 40% дітей умирають від непластичного ураження.
Для гетерозиготних носіїв синдрому Луї - Бар ризик умерти від раку в 6-8 разів вищий, ніж у популяції. Так, у матерів найбільш часто розвивається рак молочної залози, а у батьків- адено-карцинома шлунка, що необхідно враховувати при обстеженні родини та розробці тактики ведення. Часто у хворих та їх батьків виявляють ознаки остеопорозу, який розвивається вторинно на тлі непластичного ураження.
Диференційна діагностика.
Проводиться з іншими формами факоматозів та захворюваннями, що супроводжуються ангіоматозним ураженням, неврологічною симптоматикою, зокрема атактичним, мозочковим, стріо-палідарним симптомокомплексом.
Тактика ведення. Рекомендації щодо запобігання ускладнень.
Лікування неврологічних розладів симптоматичне. У комплекс терапії включають імуномодулятори, препарати, спрямовані на нормалізацію процесів репарації ДНК, для запобігання розвитку остеопорозу- препарати кальцію.
Плин захворювання неухильно прогресуючий. Діти гинуть від сепсису, пневмоній або пухлинних уражень.
Прогноз несприятливий.
Профілактика полягає в медико-генетичному консультуванні родини.
Тактика ведення повинна бути спрямована на попередження ускладнень у перебігу хвороби, на попередження пухлинного росту, зниження рівня інвалідизації та смертності хворих, на попередження розвитку пухлин у гетерозиготних носіїв патологічного гену.
Враховуючи онкогенетичний характер патології, хворим та їх батькам не показано:
інсоляція;
призначення біостимуляторів, фізіо- та гормонотерапії.
Рентгенобстеження, по можливості, замінювати УЗД, ЯМРТ та КТ.
Робота батьків дитини не повинна бути пов’язана з профшкідливостями.

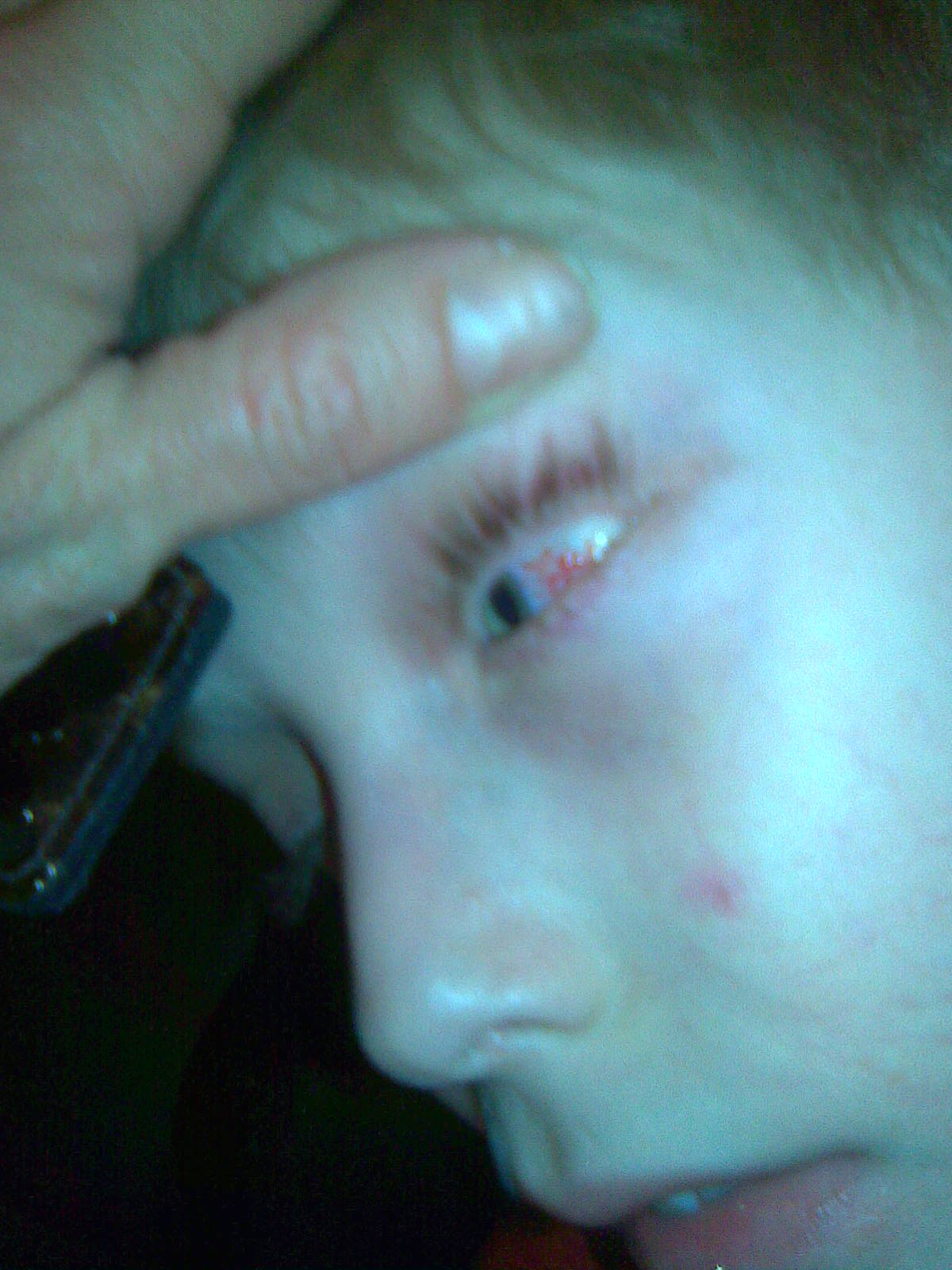
Рис.14. Синдром Луї-Бар Рис.15. Синдром Луї-Бар