

2.4 Истинная энергия активации
Из квантово-механического рассмотрения гармонического осциллятора следует, что энергия колебаний атомов в молекулах не может быть меньше ½ hv для каждого коле-бания, где v-частота колебания, h- постоянная Планка.
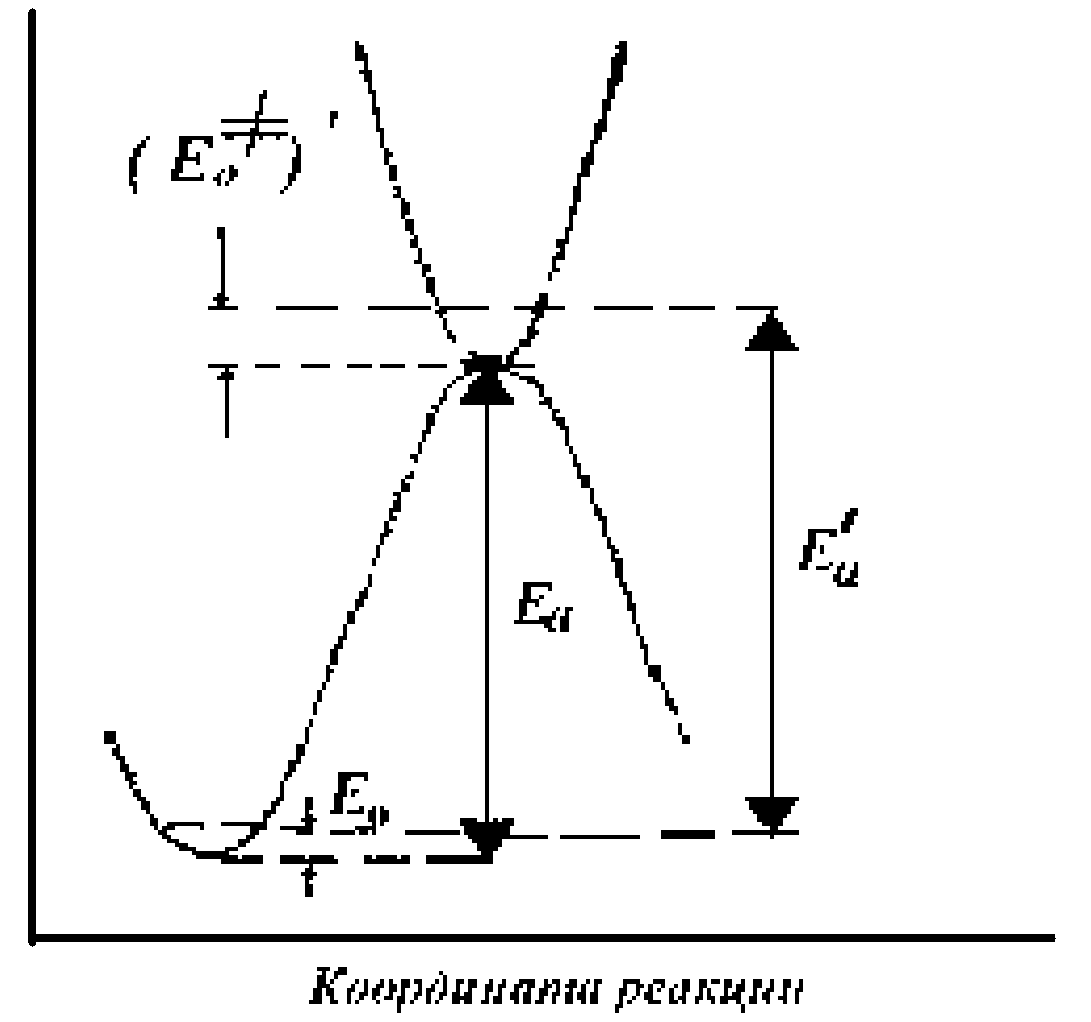
Рисунок 2.7 - Диаграмма потенциальной энергии реагирующей системы с учетом нулевых колебательных энергий исходного и переходного состояний
Поэтому полная
энергия системы не может быть меньше
нулевой колебательной энергии:
![]() ,
,
где п—число колебательных степеней свободы.
Активированный комплекс также имеет свое значение нулевой колебательной энергии (Ео≠), поскольку он находится в минимуме потенциальной энергии по всем степеням свободы, кроме поступательного движения по координате реакции.
![]()
Таким образом, полная энергия активированного комплекса не может быть меньше, чем Eo≠ = Ea + (Eo≠)´ , где (Eo≠)´ - нулевая колебательная энергия активированного комплекса (рисунок 2.7).
Истинная энергия активации Еа' равна разности нулевых колебательных энергий переходного и исходного состояний Еа'= Eo≠ -Ео.
2.5 Кинетический изотопный эффект
Кинетический изотопный эффект (КИЭ) - это изменение скорости реакции при замене некоторого атома на его изотоп. Например, рассмотрим скорости реакций ионизации нитрометана и его дейтерозамещенного аналога:

Кинетический изотопный эффект характеризуется отношением констант скоростей реакций, в которых участвуют соответствующие изотопы: КИЭ = kH/kD.
Различают первичный и вторичный изотопные эффекты. Изотопный эффект называется первичным в том случае, если изотоп входит в состав рвущейся или образующейся связи. Вторичный изотопный эффект имеет место, когда замена изотопов влияет на скорость процесса, однако сами эти изотопы не участвуют в разрыве или образовании связи в определяющей скорость стадии процесса.
Например, в реакции DCH2NО2 + OH- DCHNО2- + H2O по сравнению с ионизацией СН3NO2 наблюдается вторичный изотопный эффект.
Знание кинетического изотопного эффекта важно для понимания механизмов реакций. В частности, из величин КИЭ в реакциях разрыва и образования связей Х—Н, X—D, Х—Т можно сделать вывод, является ли эта стадия определяющей скорость или нет.
При замещении атомов на их изотопы не происходит изменений в поверхности потенциальной энергии молекулы, но изменяются средние колебательные энергии молекулы и активированного комплекса. Например, нулевые колебательные энергии молекул H2, HD, D2 соответственно составляют 25,9 кДж/моль, 22,5 кДж/моль и 18,4 кДж/моль. Энергия диссоциации молекулы Н2 равна 431 кДж/моль, a D2- 438 кДж/моль, т.е. реакция диссоциации с участием Н2 пойдет быстрее, чем с участием D2 (рис. 2.8). Аналогично, связь C—D прочнее связи С—Н на 5 кДж/моль, и т. д.
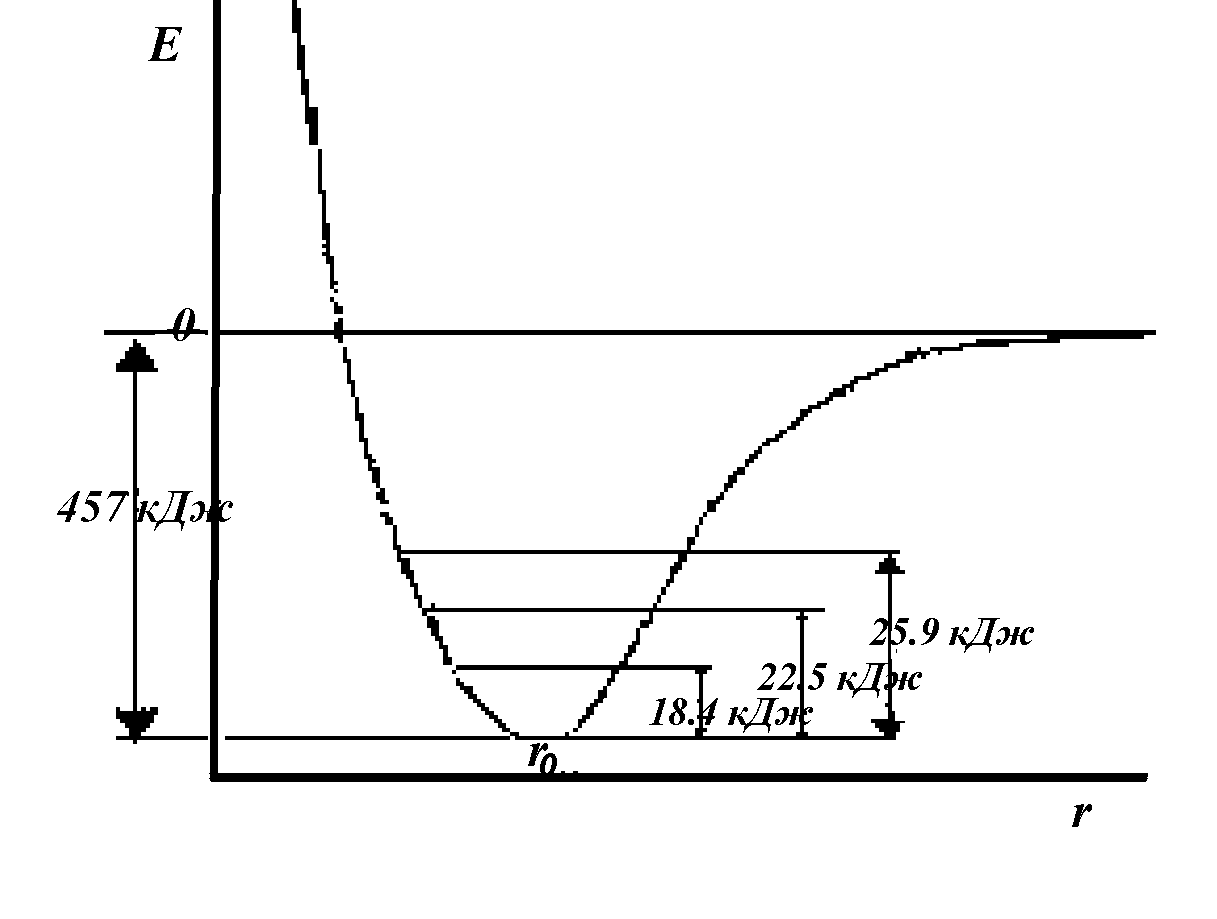
Рис. 2.8. Кривая потенциальной
энергии молекул Н2, HD и D;
со значениями нулевых коле-
бательных уровней энергии
Рассмотрим влияние замены изотопа водорода на скорость мономолекулярных реакций. Если в активированном комплексе полностью разрывается связь С—Н или С—D, то быстрее будет проходить реакция с С—Н-соединением, а для С—D-соединения энергия активации будет выше и скорость реакции меньше.
Если связь С—Н или С—D в активированном комплексе остается нетронутой, то разность нулевых колебательных уровней в переходном и исходном состояниях будет одинаковой и изотопный эффект не обнаруживается. Если в активированном комплексе связь С—Н или С—D ослаблена (рис. 2.9), то изотопный эффект будет меньше, чем для случая с полным разрывом связи.
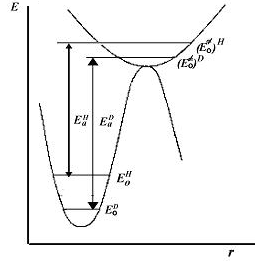
Рис. 2.9. Иллюстрация кинетического изотопного эффекта для реакции, в переходном состоянии которой ослабляется связь С—Н (С—D): Ео и Ео≠—нулевые колебательные уровни
исходного и переходного состояний; EаH и ЕаD — энергии активации с участием протия и дейтерия
Кинетический изотопный эффект проявляется не только в изменении скоростей химических реакций, но и в изменении констант химических равновесий. Так, равновесный процесс, протекающий с понижением энергии Гиббса (т.е. с образованием более стабильных продуктов), в котором происходит разрыв связи С—Н или С—D, для дейтерированных молекул сильно смещен вправо.