

1.1 Спектральні властивості та енергетична структура органічних макромолекул
Спектральні властивості та перебіг фотофізичних процесів в органічних сполуках визначаються особливостями енергетичної структури їх молекул. Зокрема, важливу роль відіграє наявність π-електронмістких систем у молекулі.
Нуклеїнові
кислоти (НК): ДНК та РНК належать до
полімерів в яких
 -
електрони локалізовані в межах
елементарної ланки, а точніше у хромофорних
групах бічного ланцюга. Розглянемо
детальніше особливості фотофізичних
процесів в системах такого типу.
-
електрони локалізовані в межах
елементарної ланки, а точніше у хромофорних
групах бічного ланцюга. Розглянемо
детальніше особливості фотофізичних
процесів в системах такого типу.
Енергетичні стани та можливі фотофізичні процеси в органічних макромолекулах можуть схематично бути представлені за допомогою діаграми Яблонського (Рис. 1.1.1).

Рис. 1.1.1 Діаграма енергетичних станів органічної молекули (діаграма Яблонського).
Основний,
перший та другий збуджені електронні
стани позначені відповідно S ,S
,S та S
та S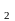 ,
а перший збуджений триплетний стан –
T
.
Кожен електронний рівень розщепляється
на серію коливальних (позначені як 0,1,2
і т.д.), а кожен коливальний в свою чергу
на серію дуже близько розташованих
обертальних рівнів
[11].
,
а перший збуджений триплетний стан –
T
.
Кожен електронний рівень розщепляється
на серію коливальних (позначені як 0,1,2
і т.д.), а кожен коливальний в свою чергу
на серію дуже близько розташованих
обертальних рівнів
[11].
Після поглинання фотона один з електронів, який належить молекулярній орбіталі молекули в основному стані, переходить на молекулярну орбіталь з більшою енергією, що відповідає переходу молекули в збуджений стан. Причому, якщо спін електрона при переході не змінюється (рис. 1.1.2), то квантове спінове число лишається рівним 0, а мультиплетність обох станів дорівнює 1. Енергетичні стани з таким набором квантових чисел називаються синглетними (S0 – основний стан, S1, S2 – збуджені синглетні стани). Якщо ж електрон зі збудженого синглетного стану може перейти безвипромінювально в інший збуджений стан зі зміною спіну на протилежний (рис. 1.1.2), то в такому випадку загальне спінове число дорівнюватиме 1, а мультиплетність відповідно – 3 (триплетний стан). Відповідно до правила Хунда триплетний стан має енергію нижчу за енергію синглетного рівня з такою ж електронною конфігурацією.
|
Рис. 1.1.2 Схематичне зображення основного синглетного, збуджених синглетного та триплетного електронних рівнів з врахуванням їх спіну. |

Згідно правил відбору існує так звана спінова заборона на переходи між станами з різною мультиплетністю. Тобто переходи синглет-синглетні та триплет-триплетні – дозволені, а синглет-триплетні – заборонені. Однак, можливі відхилення від цієї заборони за наявності слабкої взаємодії між хвильовими функціями станів з різною мультиплетністю, що реалізується через спін-орбітальну взаємодію. Саме завдяки спін-орбітальній взаємодії стає можливою інтеркомбінаційна конверсія (рис. 1.1.1) – безвипромінювальні переходи між рівнями S1 та T1.
Після поглинання молекулою фотона відбувається ряд процесів, які призводять до локалізації та деактивації електронного збудження. Деактивація збудження в молекулі може відбуватися двома шляхами: безвипромінювальним та випромінювальним. До безвипромінювальних каналів деактивації електронного збудження належать: процес релаксації молекули з верхнього на нижній коливальний рівень збудженого стану внаслідок втрати енергії при зіткненнях з іншими молекулами, внутрішня конверсія (швидка релаксація молекули з коливального рівня збудженого стану на найнижчий коливальний рівень стану S (рис. 1.1.1), час релаксації 10-12с) та інтеркомбінаційна конверсія (конверсія зі стану S в T ). Ще одним з можливих безвипромінювальних деактиваційних процесів є перенесення електронного збудження на іншу молекулу. Детально процеси перенесення енергії електронного збудження будуть розглянуті в п.1.2.
До випромінювальних каналів деактивації збудження належить люмінесценція. Люмінесценція за визначенням С.І. Вавілова (1945р.) – це випромінювання надлишкове над тепловим з тривалістю більшою ніж 10-10 с. Згідно із загальною схемою фотофізичного процесу (рис. 1.1.1) люмінесценція молекул зумовлена випромінювальними переходами S1S0 (флюоресценція, характерний час загасання 10-9 10-8с) та T1S0 (фосфоресценція, характерний час загасання 10-6 103с) [12, 13].
Варто зазначити, що флюоресцентні властивості органічних сполук визначаються не лише їх енергетичною структурою, а й ймовірністю процесів деактивації збудження та особливостями перебігу процесів, що відбуваються між поглинанням та випромінюванням (зокрема механізмом переносу енергії збудження) в - електронмістких системах [12].
Ще одним шляхом деактивації збудження є фосфоресценція, як один з можливих наслідків інтеркомбінаційної конверсії між рівнями S1 в T1, яка реалізується завдяки спін-орбітальній взаємодії. Таким чином, щоб отримати фосфоресценцію, необхідно заселити триплетний стан, що досить важко зробити через слабку смугу синглет – триплетного поглинання. Тому для його заселення використовують явище інтеркомбінаційної конверсії через синглетний рівень (рис. 1.1.1). Щодо спектрального розташування, то зазвичай спектри фосфоресценції зсунуті в більш довгохвильовий бік порівняно зі спектрами флюоресценції, адже триплетний рівень за правилом Хунда міститься нижче синглетного [13].
Варто додати, що при кімнатних температурах деактивація збудження триплетного стану T1 реалізується в основному безвипромінювальним шляхом, а не через фосфоресцентне випромінювання, оскільки даний перехід заборонений правилами відбору, а отже його ймовірність дуже мала. При низьких же температурах час життя триплетного рівня збільшується та стає достатнім для спостереження фосфоресценції [13, 14].
В окремих випадках випромінювальна деактивація збудження може реалізуватися через затриману флюоресценцію. Затримана флюоресценція поділяється на два типи [14]:
1) Е-тип (тому що вперше спостерігалася для еозину). При умові, що енергетична різниця між станами Т1 та S1 досить мала, а час життя триплетного рівня достатньо великий може відбутися зворотній перехід Т1→S1. В результаті такого процесу буде спостерігатися додатковий внесок у спектр флюоресцентного випромінювання, але з більшим часом затримки, оскільки молекули певний час перебувають в триплетному стані, що передує випромінюванню з S1. Із збільшенням температури ефективність затриманої флюоресценції такого типу значно зростає. Зазвичай такий тип флюоресценції не спостерігається в ароматичних сполуках через значну енергетичну різницю між їх синглетними та триплетними станами. Проте даний процес є дуже ефективним у фулеренах.
2) P-тип (тому, що вперше спостерігалася для пірену). Існує ймовірність зіткнення двох триплетних збуджень, в результаті чого з’явиться достатня кількість енергії для повернення однієї з молекул в синглетний стан S1. Отже анігіляція триплет-триплетного збудження призведе до появи затриманого в часі флюоресцентного випромінювання. Зазвичай час затримки флюоресценції становить половину часу життя триплетного рівня [14].
Однією з важливих характеристик флюоресцентного випромінювання є його поляризація або анізотропія. Поляризаційні вимірювання дозволяють отримати інформацію про конформацію молекул в розчиннику, їх рухливість, розміри, про наявність взаємодії між молекулами, про міграцію енергії електронного збудження та ін. Для вимірювання анізотропії флюоресценції зазвичай використовують два методи: однопроменевий (використовується один канал реєстрації випромінювання) та двопроменевий (паралельна та перпендикулярна складова одночасно реєструються в двох каналах) [13].
Теоретичні значення величини анізотропії лежать в межах -0.2≤r≤0.4. Значення r досягає максимуму (0.4) за умови, що моменти переходу поглинання та випромінювання паралельні, а мінімуму (-0.2) – перпендикулярні [14].
В природі існує ряд процесів, що призводять до зменшення анізотропії або, іншими словами, до деполяризації флюоресценції. Основні з них [13, 15]:
1) обертальна деполяризація: дипольні моменти молекули при поглинанні та при випромінюванні світла різні, за умови що молекула встигає повернутися на певний кут за час життя збудженого стану;
2) резонансна передача енергії між флюорофорами, що призводить до додаткового кутового зміщення дипольних моментів, а отже і до зменшення анізотропії.
Розглянемо детальніше вищезгадані процеси.
1) Обертальна деполяризація: якщо розчин не є жорстким середовищем, тобто його в’язкість порівняно мала, то молекули розчиненої речовини швидко обертаються і за час 1-10 нс – що є середнім часом життя збудженого стану, встигають до початку процесу випромінювання змінити орієнтацію в просторі (за умови відсутності інших каналів деактивації збудження, крім флюоресценції). За рахунок таких процесів відбувається зменшення анізотропії або навіть повна деполяризація випромінювання. Вплив вищевказаних процесів на ступінь поляризації описується формулою [13]:
 -
для вертикально поляризованого світла;
(1.1.1)
-
для вертикально поляризованого світла;
(1.1.1)
 -
для неполяризованого світла.
(1.1.2)
-
для неполяризованого світла.
(1.1.2)
Де р – ступінь поляризації, р0 – гранична поляризація (значення р у відсутності деполяризації), τ – час життя випромінювання, ρ – обертальна релаксація.
2) Результатом передачі енергії між двома молекулами флюорофору буде деполяризація, яку можна описати формулою [13]:
 ,
(1.1.3)
,
(1.1.3)
де знак мінус (-) відповідає збудженню вертикально поляризованим світлом, а знак плюс (+) – збудженню неполяризованим світлом, N – число Авогадро, 2а – діаметр молекули, с – концентрація в молях/л=М, Rс – критична відстань між диполями, на якій ймовірність перенесення енергії дорівнює ймовірності випромінювання.