

99 кругов АДА
.pdf73. Методы пренатальной диагностики. Хорион- и пластоцентез, амниоцентез, кордоцентез.
Непрямые: отбор групп высокого риска, нуждающихся в детальном наблюдении -акушерско-генекологические -медико-генетические
-биохимические (тесты на альфа-фетопротеин, эстрадиол)
Прямые:
Неизвазивные: УЗИ, электрокардиография, рентгенография и др Инвазивные:
Хорионтоцентез, биопсия проводится в сроки 8-11 недель, трансцервикально 5-20 мг ворсин хориона
Противопоказания: инфекция, многоплодная беременность Осложнения: внутриматочные течения, инфекции 2-4%
Плацентоцентез, в сроки 10-20 недель, трансабдоминально Противопоказания: миома матки, переднебрюшинное расположение петель кишечика Осложнение: кровотечение в 1-2%
Амниоцентез – взятие амниотической жидкости, на 16-20 неделях беременности, трансабдоминально Противопоказание: маловодие Осложнение: инфекция в 1% Достаточно 10-15 мл околоплодных вод
Клетки которые там есть: собственно амниоциты и эпителиальные клетки и фибробласты кожи, кишечника и ротовой полости плода
Кордоцентез – взятие пуповинной крови на 20-26 неделе беременности, трансабдоминально Осложнение: 1-2% нарушение сердечной деятельности плода
74. Наследственные болезни обмена, классификация, патогенез.
Обширный класс моногенных наследственных заболеваний, обусловленных мутациями генов, кодирующих ферменты, транспортные или сигнальные белки.
Этому классу заболеваний относят около 700 форм, и это число постоянно увеличивается. Суммарная частота наследственных болезней обмена среди новорожденных 1:3000-1:5000.
Многие формы наследственных болезней обмена протекают с поражением нервной системы.
Мутация в гене Дефекты в синтезе или катаболизме:
белков, углеводов и жиров.
Дефект в ферменте или транспортный белок, что приводит к блоку метаболического пути.
I. Нарушение промежуточного метаболизма с острой или хронической интоксикацией:
аминоацидопатии; органические ацидурии; нарушения обмена мочевины; непереносимость сахаров (галактоземия, непереносимость фруктозы)
II. Нарушение промежуточного метаболизма с дефицитом продукции или утилизации энергии: цитоплазматические и митохондриальные
III. Нарушение синтеза или катаболизма сложных молекул: лизосомные болезни; пероксисомные болезни; нарушение гликозилирования
IV. Болезни обмена нейротрансмиттеров:
некетотическая гиперглицинемия; нарушение метаболизма γ- аминомаслянной кислоты; дефицит сульфит оксидазы
Классификация НБО (наследственных болезней обмена).
1.НБО а.к. и органических кислот: альбинизм, фенилкетонурии
2.НБО углеводов: глюкозурии, (галактеземия), болезнь Гирке
3.НБО липидов: плазматических (семейная гиперхолистеренемия), клеточных (сфинголипидозы, болезнь Гоше, Фабри, Неймана-Пика)
4.НБО стероидных гормонов: врожденная гиперплазия коры надпочечников и адреногенетальный синдром
5.НБО пуринов и пиримидинов: синдром Клиглера-Нарьяра, синдром Леше-Нихана (сам себя бьет)
6.НБО гема и порфиринов: гемолитическая анемия, острая перемежающаяся порфирия
7.НБО металлов: болезнь Вильсена-Коновалова
8.Лизосомные болезни/болезни накопления: мукополисахаридозы: болезнь Краббе, болезнь Тея-Сакса
9.Пероксисомные болезни: болезнь Цельвегера, синдром Ревсумма
10.НБО соед.ткани, мышц и костей : фибродисплазия
11.Митохондриальные болезни: болезнь Кернса-Сейра, синдром Пирсона
75. Принципы диагностики наследственных болезней обмена.
I. Диагностика в доклинической стадии:
IA. Программа массового скрининга новорожденных
Фенилкетонурия
Гипотиреоз Адрено-генитальный синдром Муковисцидоз Галактоземия
II. Селективный скрининг
IIA. Отбор лиц с подозрением наследственные болезни обмена IIB. Уточняющая диагностика
76. Просеивающие программы, цели и задачи, требования к методам, используемым в программах скрининга.

Двухэтапное система обследования на НБО: 1) просеивающие программы (массовый скрининг, селективный) 2) исп методы подтверждающие диагноз
Массовый скрининг новорожденных:
Скрининг – предположительное выявление не диагностированных ранее болезней\дефекта с помощью тестов, обследований и других процедур, дающих быстрый ответ.
В практике для массового скрининга используют кровь из пяточки, высушенную на фильтровальную бумагу.
Цели и задачи МС:
Доклиническое выявление, когда раннее лечение дает хороший эффект 2)изучение распространенности аномалий 3) популяционные исследования по определению частоты метаболического дефекта 4) углуюление знаний о сущности уже известных НБО 5) разработка методов диетотерапии 6) изучение особенности бх и клинических вариантов патологии в период новорожденности и др возрастных периодах 7) выявление гетерозиготного носительства
Требования к НБО, которую целесообразно выявлять при МС:
1)Заболевания, приводящие к серьезному уменьшению качеству жизни и трудоспособности без лечения 2)заболевания, которые достаточно распространены в популяции 3) заболевания, для которых существуют методы диагностики 4) заболевания, которые поддаются лечению с достижением принципиального эффекта для больного и общества, разработаны четкие методики профилактики.
МС: фенилкетонурия (1:8000), врожденный гипотериоз 1:4000, адреногенитальный синдром, галактоземия, муковисцидоз
Требования к методам: 1) метод должен быть диагностически значимым 2) надежен и воспроизводим 3)использует легкодоступный материал 4) экономически выгодный 5) не должен давать ложноположительных результатов.
Время диагностирования – промежуток между периодом когда можно диагностировать и лечение еще эффективно
77. Селективный скрининг на наследственные болезни обмена.
Просеивание континентов повышенного риска, в котором ожидается накопление исследуемой болезни обмена. Начальные тесты с мочой,

полуколичественные и количественные тесты. Самый полезный материал для выявления возможного метаболического нарушения при нбо – хронометрированная и измерительная проба мочи. Моча собирается в течении 24 часов без консервантов.
Проба Бенедикта: [сахара], ↑ - осадок красного цвета, ↓ - осадок зеленого
Проба Легаля: уровень кетоновых тел, ацетона. Норма: розовая, ↑ - яркосиреневая
Проба Убирмейра: на наличие продуктов гниения белков (индикан) – опред.удельный вес
Проба Сурковича: [Са], в балллах 0 – гипокальциурия, 1-2 – норма, 3 – гиперкальциурия
Проба Селиванова: качественное обнаружение глюкозы – вишнево-красный
– патология фруктоземия
Проба Нечипоренко: эритроциты, лейкоциты, цилиндры: нарушение соот.показателей: беременность, патол.почек, передозировка медикаментами
Проба по Зимницкому: плотность, суточный объем, степень распределения на протяжении суток: олиго/полиурию, гипо/гиперстенурии
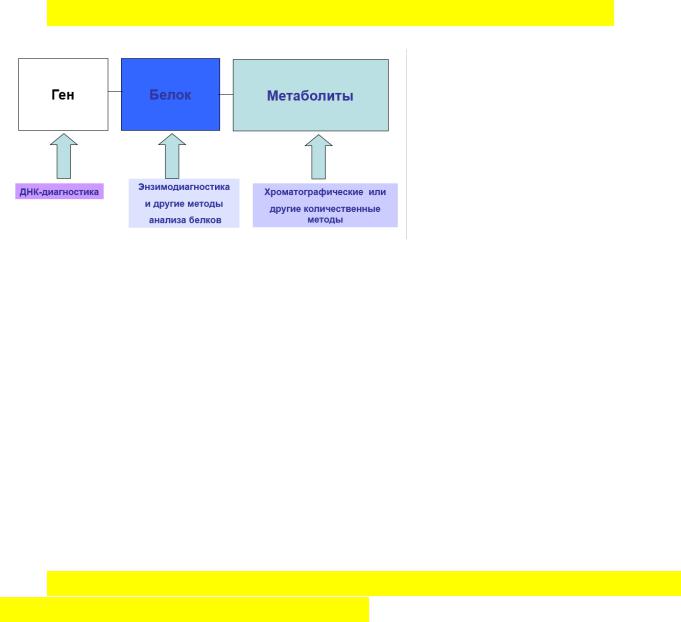
78. Методы подтверждающей диагностики наследственных болезней обмена.
Методы энзимодиагностики лизосомных и митохондриальных болезней. Тандемная масс-спектрометрия. Общие принципы: 1) для определения всех известных а\к-32 2) из одного пятна крови 4) быстро 5) проведение уже на 3 день жизни 6) низкая частота повторов 7) по принципу 1 метод-несколько болезней 8) низкая стоимость.
Методы подтверждения диагноза:
Количественный автоматический анализ аминокислот Энзимодиагностика
Тандемная масс-спектрометрия
Хромато-масс-спектрометрия
Тандемная хромато-масс-спектрометрия
Высокоэффективная жидкостная хроматография
Газовая хроматография

79. ДНК-диагностика моногенных болезней, основные методические основы: блот-гибридизация по Саузерну, ПЦР, гель-элетрофорез
Цель блот-гибридизации: выявление определенной последовательности днк в образце
1.Выделяется ДНК у любой ядросодержащей клетки (чаще это лейкоциты)
2.Тотальная ДНК обрабатывается одной определенной рестриктазой: накопление рестрикционных фрагментов разной длины
3.Ставят электрофорез в огорозном или полиакриламидном геле (ПАА)
4.На влажный гель кладут нитроцеллюлозный/нейлоновый фильр. Во влажной среде осмотическим током жидкости происходит перенос фрагментов ДНК с влажного геля на фильтр, там образуется реплика ДНК-рестрикта
5.При переносе в щелочной среде ДНК денатурирует (т.е становится одноцепочечной)
6.В таком состоянии ДНК гибридизуется с меченным ДНК-зондом на исследуемый фрагмент (перенос продолжается от 12 до 24 ч зависит от длины фрагмента)
Гибридизация – образование 2нитевых структур ДНК-ДНК или ДНК-РНК Зонд – короткий одноцепочечный фрагмент ДНК, искусственно синтезируемый
комплементарный искомой последовательности и меченный флуоресцентной или радиактивной меткой
7.Для гибридизации фильтр кладут в полиэтиленовый пакет, туда же буфер и зонд. Продолжается 12 часов и более, при температуре 68-70 градусов Цельсия
8.После гибридизации отмывают и фиксируют метку с помощью рентгеновской пленки, если метка радиоактивна или микроскопируют, если метка флуоресцента.
ПЦР – многократное копирование опр.фрагментов ДНК
Цикл:
1.Денатурация (97-98 градусов)
2.Гибридизация на специфических последовательностях (отжиг от 52-70) зависит от того каким локусом становится – отжиг 58градусов
3.Синтез – локация цепи (72)
1 цикл: 5:1:1
29 циклов: 0,5:1:1
Последний: 0,5:1:7
Гель-электрофорез – разделение и идентификация фрагментов ДНК
1.Электрофоретическая камера (кювета в которой размещается гель в которой размещается два мм буфера)
2.Буфер: трис-ацетат-ЭДТА, трис-борат-ЭДТА
3.Полимер: агароза – порошок, который получают из красных водорослей, ПАА – акриламид + бисакриламид – готовят и вливают в подложку
4.Вставляют гребенку, и остужают гель до полной кристаллизации

5.В образовавшиеся лунки наносят пробы ДНК (ДНК+1/10объема бромфеноловый синий)
6.Гель в камеру, заливают буфером
7.Подключают напряжение
8.Через 30-60 минут смотрят под УФ
80.Генеалогический метод. Сбор и генетический анализ родословных. Возможные трудности при сборе анамнезе и составлении родословной.
Клинико-генеалогический метод является основным аналитическим методом в практике медико-генетического консультирования. Он применяется в следующих целях: для установления наследственного характера заболевания, для определения типа наследования и пенетратности патологического гена, для анализа генетического сцепления, при расшифровке механизмов взаимодействия генов.
Сбор начинают от пробанда, т.е больного в карту подробно записываются сведения о больном. В дальнейшем продолжают сбор данных о сибсах пробанда, его родителях, детях и т.д. Собираются сведения, касающиеся конкретного пат.признака, но и информацию о других серьезных заболеваниях и аномалиях встречающихся среди членов семьи.
На основании собранных данных строят графическую родословную.
Необходимо получать надежные и достоверные сведения о родственниках (по возможности, в 3-4м поколении).
Целесообразно указывать девичьи фамилии женщин: особенно важно для х-сцепленных заболеваний.
Сбор родословной должен проходить в свободной непринужденной обстановке, без посторонних лиц. Можно собирать родословную по анкете, но лучше лицом к лицу, лично. Иногда данные способы комбинируются.
Анализ родословной. Целью анализа родословной является установление генетических закономерностей передачи заболевания. Правильно собранная родословная помогает ответить на след.вопросы:
Встречалось ли аналогичное заболевание у родственников больного? Обнаруживается ли в родословной другое заболевание, имеющее диагностическое или прогностическое значение?
Достаточно ли имеется данных для установления типа наследования?
81. Общие закономерности этиологии и патогенеза генных болезней
Генные болезни — разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне.
У человека описаны следующие виды генных мутаций, обусловливающие наследственные болезни: миссенс, нонсенс, сдвиг рамки считывания, делеции, вставки (инсерции), нарушения сплайсинга, увеличение числа (экспансия) тринуклеотидных повторов. Любой из этих видов мутаций может вести к наследственным болезням. Даже одна и та же генная болезнь может быть обусловлена разными мутациями.
Начало патогенеза любой генной болезни и его «ключевая точка» связаны с первичным эффектом мутантного аллеля, поэтому принципиальные звенья патогенеза генных болезней можно представить следующим образом:
мутантный аллель -» патологический первичный продукт (качественно или количественно), цепь последующих биохимических процессов -» клетки -» органы -» организм. Это и есть главная общая закономерность развития генных болезней при всём их многообразии.
В зависимости от контролируемого конкретным геном продукта и от характера нарушения его при мутации соответствующим образом развёртывается патогенез болезни на молекулярном уровне.
Если в результате мутации будет вырабатываться избыточное количество продукта, то патогенез болезни в целом будет обусловлен именно усиленной генной активностью (еще не обнаружен в конкретных формах наследственных болезней).
При другом варианте патологического эффекта мутантного гена синтезируется аномальный белок. За этим следуют нарушения той системы (клетки, органа), функции которой обеспечиваются нормальным белком (серповидно-клеточная анемия).
Третий вариант патологического эффекта мутантного аллеля — отсутствие выработки первичного продукта. Чаще всего. Нарушается тот или другой процесс из всего комплекса нормального биохимического гомеостаза. Это

выражается в накоплении токсичных продуктов-предшественников (фенилкетонурия).
Известен и четвёртый вариант первичного патологического эффекта мутантно-го аллеля — выработка уменьшенного количества нормального первичного продукта (бетта-талассемия, акаталазия). Патогенез таких заболеваний отличается большой вариабельностью, поскольку наряду с нормальным путём обмена веществ будут протекать и патологические процессы.
82. Груз наследственной патологии в мединском и социальном аспекте. Оценка тяжести медицинских и социальных последствий.
Генетический груз популяции - распространенность неблагоприятных мутаций, которые проявляются как наследственные болезни или как факторы, снижающие устойчивость (приспособленность) людей к воздействию внешней среды, другими словами, определяют предрасположенность к заболеваниям.
Известно, что вся наследственная патология определяется грузом мутаций: вновь возникающих и унаследованных из предыдущих поколений.
Регистр - метод учета достаточно важных сведений, поступающих регулярно Задача - предупреждение генетического заболевания
Цели
1.Терапевтическая цель - облегчение (по возможности) течения заболевания в последующих поколениях, ведение диспансерной работы среди больных и осуществление (по возможности) доклинической и преклинической диагностики
2.Повышение эффективности медико-генетического консультирования
3.Осуществление мониторинга по оценке результатов генетического консультирования и пренатальной диагностики
4.Научная цель - сбор и анализ эпидемиологических данных, поиски родоначальников отдельных наследственных заболеваний
5.Профилактическая цель - определение риска передачи патологического гена лицам с наследственными заболеваниями потомкам для проведения МГК и пренатальной диагностики, когда это возможно.
6.Сбор информации об ассоциации ВПР и возможных мутантных агентов
7.Цитогенетичекая цель - регистр является источником информации о взаимосвязях фенотипа пораженного индивидуума с хросомными аномалиями

8. Эпидемиологическая цель - выявление популяционной частоты наследственного заболевания; в случае мультифакториальной патологии - установление роли факторов внешней среды
83. Общая характеристика болезней аминокислотного обмена. Фенилкетонурия (дефект гена, метаболическая нарушения, клиническая картина, диагностика, лечение, профилактика)
Болезни аминокислотного обмена - группа заболеваний, обусловленных наследственными нарушениями метаболизма различных аминокислот (фенилаланина, тирозина, лизина, гистидина, глицина, триптофана, метионина), а также дефектами цикла синтеза мочевины (гипераммониемии)
Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена, локализующегося в длинном плече 12 хромосомы.
Воснове болезни лежит дефицит фермента фенилаланин -4- гидроксилазы, обеспечивающего превращение фенилаланина в тирозин. В результате метаболического блока происходит значительное накопление в тканях и жидкостях больного организма фенилаланина и таких его производных, как фенилпировиноградная, фенилмолочная, фенилуксусная кислоты, фенилэтиламин, фенилацетилглютамин, и др.
Впатогенезе ФКУ имеют значение следующие механизмы:
Прямое токсическое действие на ЦНС фенилаланина и его производных; Нарушение в обмене белков, липо- и гликопротеидов; Расстройства транспорта аминокислот; Нарушение метаболизма гормонов;
Нарушение обмена моноаминовых нейромедиаторов (катехоламинов и серотонина); Нарушение функции печени : диспротеинемия, генерализованная
гипераминоацидемия, повышение ДФА, метаболический ацидоз, нарушение окислительной и белковосинтезирующей функции клеточных органелл.
Частота классической ФКУ среди новорожденных по данным массового скрининга в среднем колеблется от 1 : 5000 до 1 : 10000 по разным регионам России.