

2.2 Формализм химической термодинамики
Продвинуться к поставленной цели поможет предположение о частичном равновесии. Пусть в системе реагентов установились все виды термодинамического равновесия (исчезли пространственные перепады давления, концентраций, температуры) и остались лишь неравновесные значения активностей реагентов. Пусть оставшиеся неуравновешенные химические компоненты претерпевают изменения в соответствии с реакциями:
ν1R1+ν2R2+…νiRi → νi+1Ri+1 + … + νkRk (2.36)
νi – стехиометрические коэффициенты. Если νi в левой части уравнения считать отрицательными, а в правой положительными, то из (2.36) получим:
K
Σ νiRi = 0, это - стехиометрическое уравнение реакции (2.37).
i=1
Пусть μi- молекулярная масса i-того – компонента.
Естественно считать, что для каждой конкретной реакции типа (2.37) должен существовать свой, характерный для данной реакции, параметр, ξ, определяющий её состояние на пути протекания реакции для массы реагентов:
m i
– mio
= νiμiξ
i
– mio
= νiμiξ
…………………
mk – mko = νkμkξ (2.38)
или nj – nio = νiξ для числа молей
Здесь ξ ≡ координата реакции, степень её полноты 0 ≤ ξ ≤ 1
![]()
![]() При
ξ = 1 (один шаг реакции) прошел один
эквивалент реакции. Складывая (2.38) и,
помня о законе сохранения массы, получим
для изолированной системы реагентов:
При
ξ = 1 (один шаг реакции) прошел один
эквивалент реакции. Складывая (2.38) и,
помня о законе сохранения массы, получим
для изолированной системы реагентов:
Это - стехиометрическое соотношение. ξ наряду с Р, Т при заданном mio полностью характеризует состояние системы и, следовательно, является термодинамическим параметром.
Пример: H2O = OH-+ H+
n1 = n10 – ξ n2 = n3 = ξ,
т.е. ξ – это степень диссоциации в химическом смысле.
Дифференцирование (2.38) по времени дает:
и![]() ли
ли
![]()
![]()
![]()
![]()
Е![]()
![]()
![]() сли
в системе протекает r
различных реакций типа (2.37), то появится
дополнительный индекс ρ – номер
реакции:
сли
в системе протекает r
различных реакций типа (2.37), то появится
дополнительный индекс ρ – номер
реакции:
З![]() десь
ξρ – координата, Vρ
– скорость ρ-той реакции.
десь
ξρ – координата, Vρ
– скорость ρ-той реакции.
Сколько реакций линейно независимых, т.е. сколько степеней свободы ξρ у системы? Ответ на это дает критерий Жуге: число независимых переменных равно рангу матрицы, составленной из стехиометрических коэффициентов ║νiρ║.
Вернемся к системе, в которой имеются и химические и термодинамические степени свободы: (Т, P, ni) – могут изменяться, но одновременно по всей системе – так называемое частичное равновесие. Рассмотрим сначала случай одной реакции. Пусть термодинамические потенциалы являются функцией, например T, V, ni, или T, V, ξ при заданном nio.
Объединим 1 и 2 начала термодинамики для закрытой системы.Из (2.22):
![]()
![]() δQ/
= TdS – δQ
(2.44),
а δQ
выразим из (2.17): δQ=dU+pdV
и подставим в (2.27):
δQ/
= TdS – δQ
(2.44),
а δQ
выразим из (2.17): δQ=dU+pdV
и подставим в (2.27):
![]()
![]() Будем
считать S=S
(T, V, ξ),
тогда:
Будем
считать S=S
(T, V, ξ),
тогда:
а![]()
![]() налогично,
U=U(T,
V, ξ):
налогично,
U=U(T,
V, ξ):
Т![]()
![]() еперь
из (2.45):
еперь
из (2.45):
Независимые переменные dT и dV могут иметь любой знак, а ∂Q/ всегда ≥ 0. Это значит, что выражения в скобках при dT и dV равны нулю.
Т![]() аким
образом:
аким
образом:
![]()
![]() Этот
же результат можно получить короче,
непосредственно используя уравнение
Гиббса (если читатель его помнит):
Этот
же результат можно получить короче,
непосредственно используя уравнение
Гиббса (если читатель его помнит):
г![]() де
μi –
химический потенциал i-того
реагента (не путать с молекулярной
массой в (2.38-2.42)!).
де
μi –
химический потенциал i-того
реагента (не путать с молекулярной
массой в (2.38-2.42)!).
![]()
![]() Сравнивая
это выражение с выражением для dU
из (2.45), получим формулу (2.45) в несколько
более общем виде:
Сравнивая
это выражение с выражением для dU
из (2.45), получим формулу (2.45) в несколько
более общем виде:
![]() Производные
от потенциалов по координате называются
химическим сродством реакции.
Обозначая его буквой А, получим из
(2.49) или (2.51):
Производные
от потенциалов по координате называются
химическим сродством реакции.
Обозначая его буквой А, получим из
(2.49) или (2.51):
г![]() де
V – скорость реакции.
де
V – скорость реакции.
Это важное соотношение называется неравенство де-Донде (1937 г.).
В чем его интерес:
Если А>0, то V>0
Если А<0, то V<0
Если А=0, то AV=0, т.е. реакция обратима, а, значит, V=0. Итак, V имеет тот же знак, что и А и обращается в 0 тогда, когда А=0. Обратное же, вообще говоря, не верно: V=0 может означать истинное равновесие, когда А=0, но может и ложное, когда А≠0, а реакция не идет из-за кинетических ограничений (без катализатора, фермента).
Итак, А=0 является необходимым и достаточным условием истинного равновесия. А(Т,V,ξ)=0 – уравнение, определяющее некую поверхность ξ=ξ(TV) на которой имеется равновесие, и которая отделяет область с А<0, от области А>0 (рис. 2.4).
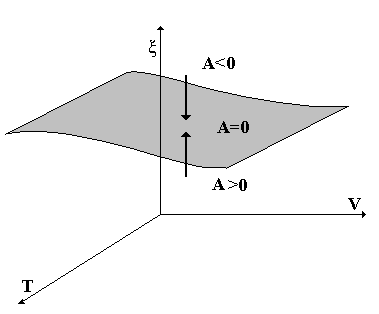
Рис. 2.4
Но особую ценность неравенство де-Донде приобретает для случая нескольких одновременных в одном месте реакций:
П![]()
![]()
![]()
![]() ри
этом, используя (2.24) получим
ри
этом, используя (2.24) получим
Аρ – сродство ρ-той реакции.
Здесь неравенство относится лишь к сумме! И отсюда возникает принципиально новый момент.
О![]() тдельные
слагаемые суммы AρVρ
могут теперь иметь отрицательный знак!
Но AρVρ<0
означает, что ρ-тая реакция в данной
системе идет в сторону, противоположную
той, которую ей диктует её собственное
(невозмущенное сопряжением) сродство,
т.е. куда она шла бы, если бы была
изолированной. Здесь нет противоречия
со вторым началом термодинамики.
Положительность полной функции диссипации
обеспечивается при этом большой величиной
σρ «сопрягающих» реакций.
Например для r=2:
A1V1+A2V2≥0.
Пусть │A1V1│>│A2V2│,
тогда A2V2
может быть <0, но при этом появляется
ограничение на скорость сопряженной
реакции:
тдельные
слагаемые суммы AρVρ
могут теперь иметь отрицательный знак!
Но AρVρ<0
означает, что ρ-тая реакция в данной
системе идет в сторону, противоположную
той, которую ей диктует её собственное
(невозмущенное сопряжением) сродство,
т.е. куда она шла бы, если бы была
изолированной. Здесь нет противоречия
со вторым началом термодинамики.
Положительность полной функции диссипации
обеспечивается при этом большой величиной
σρ «сопрягающих» реакций.
Например для r=2:
A1V1+A2V2≥0.
Пусть │A1V1│>│A2V2│,
тогда A2V2
может быть <0, но при этом появляется
ограничение на скорость сопряженной
реакции:
Р![]() еакция
1 называется сопрягающей, 2 – сопряженной.
еакция
1 называется сопрягающей, 2 – сопряженной.
Таким образом Аρ приобретает важное значение для анализа Vρ .
Физический смысл химического средства ρ – той реакции достаточно прост: это мера скорости падения свободной энергии системы реагентов в процессе протекания ρ-той реакции. Численно его значение совпадает с изменением свободной энергии реакции, когда она продвигается на один шаг (ξ = 1), т.е. когда прореагируют стехиометрические количества реагентов. Но знак его по определению противоположен: чем сильнее убывает свободная энергия системы, тем более положительна сила, движущая процесс, т.е. Аρ .
В общем случае, для любого числа независимых реакций
![]() (2.56)
(2.56)
![]()
![]() Условия
истинного равновесия:
Условия
истинного равновесия:
Это соотношение не следует путать с формулой (2.39), они имеют совершенно разный смысл.
![]()
![]() Значение
Аρ, как и ∆Gρ
, выражается через стандартную часть,
соответствующую превращению реагентов
при стандартных условиях (нормальном
давлении, комнатной температуре и
активности реагентов, равных 1 м/л) и
добавку, учитывающую отличие активностей
от стандартных значений:
Значение
Аρ, как и ∆Gρ
, выражается через стандартную часть,
соответствующую превращению реагентов
при стандартных условиях (нормальном
давлении, комнатной температуре и
активности реагентов, равных 1 м/л) и
добавку, учитывающую отличие активностей
от стандартных значений:
Эта формула наглядно отражает очевидный факт: чем больше активность субстратов реакции, входящих в левую часть уравнения (2.30) (т.е. имеющих отрицательные νρi), тем больше сродство.
Первый член в (2.58), энтальпийный по своей природе, отражает собственную энергетику акта взаимодействия реагентов, а второй, энтропийный, отвечает за вероятность встречи реагентов.
![]()
![]() Подставляя
(2.58) в условия равновесия реакций (2.57),
получим:
Подставляя
(2.58) в условия равновесия реакций (2.57),
получим:
Т![]()
![]()
![]() .к.
константа равновесия по определению
.к.
константа равновесия по определению
![]()
![]()
т![]() о
о
где μoj – стандартная часть химического потенциала j- того реагента:
Эти формулы пригодятся нам в дальнейшем.
Итак, соотношение де-Донде (2.55) указывает на принципиальную возможность термодинамического сопряжения двух реакций, т.е. на обращение скорости сопряженной реакции за счет большого сродства сопрягающей реакции. Действительно, в живых системах протекание процессов против их естественного сродства это типичное явление, делающее возможным возникновение и поддержание устойчивого неравновесия в целом ряде важных клеточных систем за счет сопряжения их со спонтанно протекающими процессами. Множество примеров мы рассмотрим в дальнейшем. Соотношение де-Донде показывает, что в этом нет противоречия со вторым началом термодинамики. Посмотрим, однако, что говорит о такой возможности формальная химическая кинетика, основанная на законе действующих масс.
Рассмотрим пример простейшей химической реакции превращения веществ R1 и R2 в R3.
![]()
![]()
![]()
![]() Её
скорость V=dn3/dt
(n – число молей) это
разность скоростей прямой и обратной
реакции:
Её
скорость V=dn3/dt
(n – число молей) это
разность скоростей прямой и обратной
реакции:
г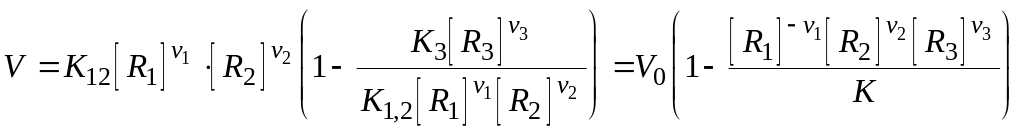
![]() де
К1,2 и К2 контрасты
скоростей прямой и обратной реакции,
[Ri]
– активности реагентов а ν1,
ν2, ν3,
положительные числа. Преобразуем (2.64)
де
К1,2 и К2 контрасты
скоростей прямой и обратной реакции,
[Ri]
– активности реагентов а ν1,
ν2, ν3,
положительные числа. Преобразуем (2.64)
Здесь К – константа равновесия, равная К1,2/К3.
![]()
![]() С
другой стороны K можно
связать с А по (2.61) и (2.58):
С
другой стороны K можно
связать с А по (2.61) и (2.58):
и получить из (2.66):
V=V0(1-e-A/RT) (2.67)
Это общее выражение можно получить для любого другого вида реакций. О чем оно говорит? О том, что знак V всегда совпадает со знаком А! Иначе говоря, термодинамика разрешает сопряженной реакции идти против её сродства, а кинетика не позволяет этого. Парадокс?! Нет. Просто условие возможности сопряжения (2.55) являются необходимым, но не достаточным для реального сопряжения. Чтобы сопряжение в смысле изменения направления сопрягаемой реакции действительно произошло необходим специальный физический механизм изменения знака сродства А2 за счет части сродства А1, передаваемой из первой реакции во вторую.
Возможны 2 типа механизмов обеспечивающих такое взаимодействие.
Энергетический механизм. Это передача части уменьшения свободной энергии системы при протекании первой реакции реагентам второй в виде какого-то вида работы над ними, увеличивающей их внутреннюю энергию или энтальпию, т.е. стандартную часть средства, А20. Это может быть деформация реакционного комплекса реагентов второй реакции, создание электрического поля, облегчающего взаимодействие реагентов и т.п. Макромолекулы ферментов, осуществляющих сопряжение, могут реализовать такие процессы.
Вероятностный (энтропийный) механизм. Это затрата части реализуемой свободной энергии первой реакции на изменение активности реагентов второй реакции, т.е. увеличение второго, этропийного члена в сродстве А2. Тривиальным примером является увеличение концентрации реагентов второй реакции, если обе реакции имеют общий продукт. Менее тривиальным, но более интересным примером является изменение при протекании первой реакции коэффициентов активности второй. Так, первая реакция может так изменить конфигурацию или свойства среды (поверхности), где протекает вторая реакция, что при той же концентрации её реагентов они станут более подвижными (например, десорбируются с поверхности) и увеличат вероятность своего участия в реакционном акте.