

-
Результаты расчета трехатомных молекул
|
2,56 А0 |
|


|
|
|
|
|



HyperChem log start -- Mon Nov 12 15:16:13 2018.
Geometry optimization, SemiEmpirical, molecule = (Гидрид бериллия).
MNDO
PolakRibiere optimizer
Convergence limit = 0.0100000 Iteration limit = 50
Accelerate convergence = YES
Optimization algorithm = Polak-Ribiere
Criterion of RMS gradient = 0.0010 kcal/(A mol) Maximum cycles = 45
RHF Calculation:
Singlet state calculation
Number of electrons = 4
Number of Double Occupied Levels = 2
Charge on the System = 0
Total Orbitals = 6
Starting MNDO calculation with 6 orbitals
E=-175.5871 kcal/mol Grad=0.000 Conv=YES(1 cycles 12 points) [Iter=1 Diff=0.00000]
Eigenvalues (eV) and Eigenvectors
|
Mol. Orbital |
1 |
2 |
3 |
4 |
5 |
6 |
|
Symmetry: |
1 SIG |
1 SIU |
1 PIU |
1 PIU |
2 SIG |
2 SIU |
|
Eigenvalue |
-13.83192 |
-12.26659 |
2.51386 |
2.51386 |
2.98640 |
7.16665 |
|
|
|
|
|
|
|
|
|
S Be 2 |
-0.67273 |
-0.00000 |
0.00000 |
0.00000 |
0.73989 |
-0.00000 |
|
Px Be 2 |
0.00000 |
0.00000 |
0.99989 |
0.01486 |
-0.00000 |
-0.00000 |
|
Py Be 2 |
0.00000 |
-0.54167 |
0.00000 |
-0.00000 |
0.00000 |
0.84059 |
|
Pz Be 2 |
-0.00000 |
0.00000 |
0.01486 |
-0.99989 |
0.00000 |
-0.00000 |
|
S H 1 |
-0.52318 |
0.59439 |
-0.00000 |
-0.00000 |
-0.47569 |
0.38302 |
|
S H 3 |
-0.52318 |
-0.59439 |
-0.00000 |
-0.00000 |
-0.47569 |
-0.38302 |
ENERGIES AND GRADIENT
Total Energy = -1282.9071321 (kcal/mol)
Total Energy = -2.044442398 (a.u.)
Binding Energy = -175.5870951 (kcal/mol)
Isolated Atomic Energy = -1107.3200370 (kcal/mol)
Electronic Energy = -2225.8062553 (kcal/mol)
Core-Core Interaction = 942.8991232 (kcal/mol)
Heat of Formation = 5.5769049 (kcal/mol)
Gradient = 0.0000014 (kcal/mol/Ang)
MOLECULAR POINT GROUP
D*H
EIGENVALUES(eV)
Symmetry: 1 SIG 1 SIU 1 PIU 1 PIU 2 SIG
Eigenvalue: -13.831922 -12.266585 2.513861 2.513861 2.986403
Symmetry: 2 SIU
Eigenvalue: 7.166646
ATOMIC ORBITAL ELECTRON POPULATIONS
AO: 2 S Be 2 Px Be 2 Py Be 2 Pz Be 1 S H
0.905139 0.000000 0.586820 0.000000 1.254020
AO: 3 S H
1.254020
NET CHARGES AND COORDINATES
Atom Z Charge Coordinates(Angstrom) Mass
x y z
2 4 0.508040 -0.43731 0.10431 -0.00000 9.01200
1 1 -0.254020 -0.43731 -1.17356 0.00000 1.00800
3 1 -0.254020 -0.43731 1.38219 -0.00000 1.00800
ATOMIC GRADIENTS
Atom Z Gradients(kcal/mol/Angstrom)
x y z
2 4 0.00000 -0.00000 -0.00000
1 1 -0.00000 0.00000 0.00000
3 1 -0.00000 -0.00000 0.00000
Dipole (Debyes) x y z Total
Point-Chg. 0.000 -0.000 -0.000 0.000
sp Hybrid 0.000 -0.000 -0.000 0.000
pd Hybrid 0.000 0.000 0.000 0.000
Sum 0.000 -0.000 -0.000 0.000
HyperChem log stop -- Mon Nov 12 15:16:39 2018
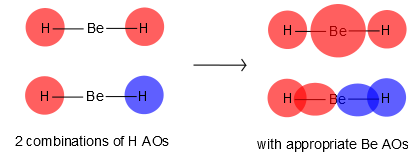
|
1σg (связ) |
|||
|
|
|
|
|
|
1σu (связ) |
|||
|
|
|
||
|
2 орбитали 1πu (несвяз)
|
|||
|
|
|
||





