

1.5.1. Основные типы термодинамических потенциалов
С учетом того, что внутренняя энергия и энтропия, наряду с общими свойствами экстенсивных параметров, обладают еще и особыми индивидуальными свойствами, вытекающими из законов термодинамики, вышеприведенное обобщенное определение термодинамических потенциалов позволяет в явном виде определить два наиболее важные с практической точки зрения термодинамических потенциала – свободные энергии Гельмгольца и Гиббса, являющиеся характеристическими функциями состояния.
А. Свободная энергия Гельмгольца
Свободная энергия Гельмгольца
F(Т,V,Nk),
как результат преобразования Лежандра
функции U(S,V,Nk),
в которой S заменена
соответствующей частной производной![]() ,
является характеристической функцией
одного интенсивного (Т) и двух
экстенсивных (V,Nk)
параметров, и в соответствии с обобщенным
определением термодинамического
потенциала в интегральной (
,
является характеристической функцией
одного интенсивного (Т) и двух
экстенсивных (V,Nk)
параметров, и в соответствии с обобщенным
определением термодинамического
потенциала в интегральной (![]() )
и дифференциальной (
)
и дифференциальной (![]() )
форме для многокомпонентной системы
определяется следующим образом:
F(T,V,N)=U-TS=-TS-PV+
)
форме для многокомпонентной системы
определяется следующим образом:
F(T,V,N)=U-TS=-TS-PV+![]() и
и
![]() .
Первые частные производные F
по интенсивным параметрам дают
экстенсивные параметры:
.
Первые частные производные F
по интенсивным параметрам дают
экстенсивные параметры:
![]() ,
а по экстенсивным – интенсивные:
,
а по экстенсивным – интенсивные:
![]() (термическое уравнение состояния),
(термическое уравнение состояния),
![]() .
В закрытых системах уменьшение свободной
энергии Гельмгольца равно работе,
совершенной системой в изотермическом
обратимом процессе. Как термодинамический
потенциал свободная энергия Гельмгольца
очень полезна при контролируемом объеме,
т.е. в газообразных системах, а также
при расчетах методами статистической
термодинамики: F=-kBlnZ,
где kB
– константа Больцмана, Z
- статистическая сумма (интеграл) по
состояниям.
.
В закрытых системах уменьшение свободной
энергии Гельмгольца равно работе,
совершенной системой в изотермическом
обратимом процессе. Как термодинамический
потенциал свободная энергия Гельмгольца
очень полезна при контролируемом объеме,
т.е. в газообразных системах, а также
при расчетах методами статистической
термодинамики: F=-kBlnZ,
где kB
– константа Больцмана, Z
- статистическая сумма (интеграл) по
состояниям.
Б. Свободная энергия Гиббса и химический потенциал.
Свободная энергия Гиббса
G(T,р,Nk),
как результат преобразования Лежандра
функции U(S,V,Nk),
в которой S и V
заменены соответствующими частными
производными
![]() и
и
![]() ,
является характеристической функцией
двух интенсивных (Т и р) и одного
экстенсивного (Nk)
параметров и в соответствии с обобщенным
определением термодинамического
потенциала в интегральной (
,
является характеристической функцией
двух интенсивных (Т и р) и одного
экстенсивного (Nk)
параметров и в соответствии с обобщенным
определением термодинамического
потенциала в интегральной (![]() )
и дифференциальной (
)
и дифференциальной (![]() )
форме определяется для многокомпонентных
систем следующим образом: G(T,p,N)=U+pV-TS=
-TS+pV+
)
форме определяется для многокомпонентных
систем следующим образом: G(T,p,N)=U+pV-TS=
-TS+pV+![]() и
и
![]() .
Для закрытых систем уменьшение свободной
энергии Гиббса равно работе, совершенной
системой в изобарно-изотермическом
обратимом процессе. Ее частные производные:
.
Для закрытых систем уменьшение свободной
энергии Гиббса равно работе, совершенной
системой в изобарно-изотермическом
обратимом процессе. Ее частные производные:
![]() ,
,![]() (термическое
уравнение состояния) и
(термическое
уравнение состояния) и
![]() -
химический потенциал, равный для
однокомпонентных систем мольной
свободной энергии Гиббса. Свободная
энергия Гиббса и химический потенциал
из всех термодинамических потенциалов
находят наиболее широкое практическое
применение при описании конденсированных
систем, так как все независимые переменные
в них (интенсивные р и Т, а также
экстенсивный параметр - число молей
компонентов Ni)
легко измеряются и контролируются.
Функция G(Т,р,Nk)
часто используется в классической
химической термодинамике в виде средней
мольной величины:
-
химический потенциал, равный для
однокомпонентных систем мольной
свободной энергии Гиббса. Свободная
энергия Гиббса и химический потенциал
из всех термодинамических потенциалов
находят наиболее широкое практическое
применение при описании конденсированных
систем, так как все независимые переменные
в них (интенсивные р и Т, а также
экстенсивный параметр - число молей
компонентов Ni)
легко измеряются и контролируются.
Функция G(Т,р,Nk)
часто используется в классической
химической термодинамике в виде средней
мольной величины: ,
которая в дифференциальной форме
записывается как функция мольных долей
компонентов хi:
,
которая в дифференциальной форме
записывается как функция мольных долей
компонентов хi:
![]() .
Последнее уравнение формально аналогично
уравнению для dG,
выведенное из обобщенного определения
G:
.
Последнее уравнение формально аналогично
уравнению для dG,
выведенное из обобщенного определения
G:
![]() .
Следовательно, средняя мольная величина
G* является
характеристической функцией, т.е. она
содержит все термодинамические сведения
о данной системе. Так, ее частные
производные по интенсивным параметрам
(р,Т) дают среднюю мольную энтропию
и средний мольный объем:
.
Следовательно, средняя мольная величина
G* является
характеристической функцией, т.е. она
содержит все термодинамические сведения
о данной системе. Так, ее частные
производные по интенсивным параметрам
(р,Т) дают среднюю мольную энтропию
и средний мольный объем:
![]() ,
,![]() .
Однако, частная производная средней
мольной величины G*
по мольной доле компонентов
.
Однако, частная производная средней
мольной величины G*
по мольной доле компонентов![]() не является химическим потенциалом,
в отличие от частного производного
свободной энергии Гиббса G(Т,р,Nk)
по числу молей компонентов:
не является химическим потенциалом,
в отличие от частного производного
свободной энергии Гиббса G(Т,р,Nk)
по числу молей компонентов:
![]() ,
которое является химическим потенциалом
компонентов
,
которое является химическим потенциалом
компонентов
![]() .
При этом
.
При этом
![]() ,
а
,
а
![]() .
.
Химический потенциал как
парциальная мольная
![]() (для многокомпонентных систем) или
просто мольная g (для
однокомпонентных систем) свободная
энергия Гиббса связан с температурой
и парциальными мольными величинами
функций состояния S
и H:
(для многокомпонентных систем) или
просто мольная g (для
однокомпонентных систем) свободная
энергия Гиббса связан с температурой
и парциальными мольными величинами
функций состояния S
и H:
![]() и
и![]() аналогичными соотношениями:
аналогичными соотношениями:
![]() ;
;
![]() .
.
Уравнение Гиббса-Дюгема с использованием частных производных как свободной энергии Гиббса G, так и ее средней мольной величины G* по интенсивным переменным, а также частных производных химического потенциала по числу молей и мольным долям формально имеет аналогичный вид для общих и изобарно-изотермических условий или процессов соответственно:
![]() и
и![]() ;
;
;
;
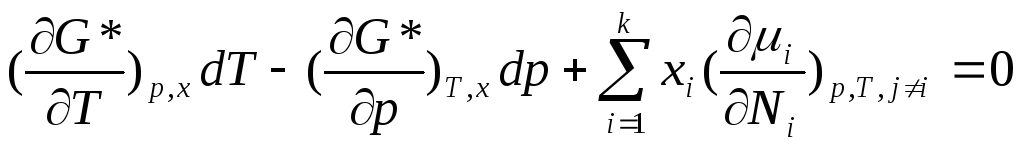 и
и
![]() .
.
В.Энтальпия
Преобразованием Лежандра функции
U(S,V,Nk),
в которой V заменена
соответствующей частной производной
![]() ,
качестве термодинамического потенциала
может быть введена также энтальпия
Н(S,р,Nk).
Она является функцией одного интенсивного
(р) и двух экстенсивных (S,Nk)
параметров. В интегральной и дифференциальной
форме уравнения для энтальпии записываются
следующим образом: Н=U+pV
и
,
качестве термодинамического потенциала
может быть введена также энтальпия
Н(S,р,Nk).
Она является функцией одного интенсивного
(р) и двух экстенсивных (S,Nk)
параметров. В интегральной и дифференциальной
форме уравнения для энтальпии записываются
следующим образом: Н=U+pV
и
![]() ,
а ее частные производные дают:
,
а ее частные производные дают:
![]() ;
;
![]() ;
;![]() .
Однако, применение энтальпии как
термодинамического потенциала не
удобно, так как функция Н(S,р,Nk)
содержит энтропию в качестве независимой
переменной, которую невозможно
контролировать. Поэтому в классической
термодинамике энтальпию, по аналогии
с внутренней энергией, обычно представляют
(в общем случае для многокомпонентной
системы) как функцию двух интенсивных
(T,р) и одной
экстенсивной (N)
переменных, которая не является
характеристической функцией, так как
изменение этой функции (ее полный
дифференциал) представляет собой
дифференциальное уравнение первого
порядка в частных производных:
.
Однако, применение энтальпии как
термодинамического потенциала не
удобно, так как функция Н(S,р,Nk)
содержит энтропию в качестве независимой
переменной, которую невозможно
контролировать. Поэтому в классической
термодинамике энтальпию, по аналогии
с внутренней энергией, обычно представляют
(в общем случае для многокомпонентной
системы) как функцию двух интенсивных
(T,р) и одной
экстенсивной (N)
переменных, которая не является
характеристической функцией, так как
изменение этой функции (ее полный
дифференциал) представляет собой
дифференциальное уравнение первого
порядка в частных производных:
![]() ,
решение которого содержит произвольную
функцию. Следовательно, энтальпия не
является в этом случае термодинамическим
потенциалом, и, соответственно, частная
производная
,
решение которого содержит произвольную
функцию. Следовательно, энтальпия не
является в этом случае термодинамическим
потенциалом, и, соответственно, частная
производная
![]() не
является химическим потенциалом. При
этом частная производная
не
является химическим потенциалом. При
этом частная производная
![]() соответствует практически очень важному
параметру - мольной теплоемкости при
постоянном давлении
соответствует практически очень важному
параметру - мольной теплоемкости при
постоянном давлении![]() ,
поэтому энтальпия как функция Н(T,р,N)
широко используется для анализа тепловых
эффектов в различных процессах. Энтальпия
впервые была введена и наиболее широко
применяется при анализе теплоты
,
поэтому энтальпия как функция Н(T,р,N)
широко используется для анализа тепловых
эффектов в различных процессах. Энтальпия
впервые была введена и наиболее широко
применяется при анализе теплоты
![]() ,
выделяемой или поглощаемой в равновесных
обратимых процессах (химических реакциях
и фазовых переходах) в закрытых системах
при постоянном давлении и составе. Было
установлено, что эта теплота обусловлена
изменением внутренней энергии и работой,
совершаемой за счет изменения объема
системы при ее переходе из начального
в конечное состояние:
,
выделяемой или поглощаемой в равновесных
обратимых процессах (химических реакциях
и фазовых переходах) в закрытых системах
при постоянном давлении и составе. Было
установлено, что эта теплота обусловлена
изменением внутренней энергии и работой,
совершаемой за счет изменения объема
системы при ее переходе из начального
в конечное состояние:
 .
Величина
.
Величина
![]() представляет собой разницу величин:
представляет собой разницу величин:
![]() ,
не зависящих от пути, по которому они
были достигнуты в закрытой систем.
Следовательно, Н=
,
не зависящих от пути, по которому они
были достигнуты в закрытой систем.
Следовательно, Н=![]() является функцией состояния,
определяемой независимыми переменными
р и Т, и равна внутренней энергии
за вычетом работы, произведенной системой
против внешнего давления при увеличении
ее объема:
является функцией состояния,
определяемой независимыми переменными
р и Т, и равна внутренней энергии
за вычетом работы, произведенной системой
против внешнего давления при увеличении
ее объема:
![]() .
Так как в конденсированном состоянии
изменение объема при любых превращениях
в изобарных условиях при обычном
(нормальном) давлении очень малы, и
работа, совершаемая при этом также мала,
изменение энтальпии в таких условиях
практически равно изменению внутренней
энергии.
.
Так как в конденсированном состоянии
изменение объема при любых превращениях
в изобарных условиях при обычном
(нормальном) давлении очень малы, и
работа, совершаемая при этом также мала,
изменение энтальпии в таких условиях
практически равно изменению внутренней
энергии.
Величина
![]() ,т.е.
изменение энтальпии в закрытой системе
при постоянном давлении равно тепловому
эффекту соответствующего равновесного
обратимого процесса (смешения, химических
реакций, фазового перехода) и характеризует
тепловой эффект или энтальпию процесса,
причем для экзотермических процессов,
сопровождающихся выделением тепла
(положительный тепловой эффект), энтальпия
отрицательная, а для эндотермических,
протекающих с поглощением тепла
(отрицательный тепловой эффект), энтальпия
положительная. Изменение энтальпии,
как и внутренней энергии, в равновесных
обратимых процессах обычно определяют
относительно некоторого стандартного
состояния.
,т.е.
изменение энтальпии в закрытой системе
при постоянном давлении равно тепловому
эффекту соответствующего равновесного
обратимого процесса (смешения, химических
реакций, фазового перехода) и характеризует
тепловой эффект или энтальпию процесса,
причем для экзотермических процессов,
сопровождающихся выделением тепла
(положительный тепловой эффект), энтальпия
отрицательная, а для эндотермических,
протекающих с поглощением тепла
(отрицательный тепловой эффект), энтальпия
положительная. Изменение энтальпии,
как и внутренней энергии, в равновесных
обратимых процессах обычно определяют
относительно некоторого стандартного
состояния.