

Книги фарма 2 / Bertram G. Katzung-Basic & Clinical Pharmacology(9th Edition)
.pdfIndomethacin
Indomethacin, introduced in 1963, is an indole derivative (Figure 36–1). It is a potent nonselective COX inhibitor and may also inhibit phospholipase A and C, reduce neutrophil migration, and decrease T cell and B cell proliferation. Probenecid prolongs indomethacin's half-life by inhibiting both renal and biliary clearance.
Clinical Uses
Indomethacin enjoys the usual indications for use in rheumatic conditions and is particularly popular for gout and ankylosing spondylitis. In addition, it has been used to treat patent ductus arteriosus. Indomethacin has been tried in numerous small or uncontrolled trials for many conditions, including Sweet's syndrome, juvenile rheumatoid arthritis, pleurisy, nephrotic syndrome, diabetes insipidus, urticarial vasculitis, postepisiotomy pain, and prophylaxis of heterotopic ossification in arthroplasty, and many others. An ophthalmic preparation seems to be efficacious for conjunctival inflammation (alone and in combination with gentamicin) to reduce pain after traumatic corneal abrasion. Gingival inflammation is reduced after administration of indomethacin oral rinse. Epidural injections produce a degree of pain relief similar to that achieved with methylprednisolone in postlaminectomy syndrome.
Adverse Effects
At higher dosages, at least a third of patients have reactions to indomethacin requiring discontinuance. The gastrointestinal effects may include abdominal pain, diarrhea, gastrointestinal hemorrhage, and pancreatitis. Headache is experienced by 15–25% of patients and may be associated with dizziness, confusion, and depression. Rarely, psychosis with hallucinations has been reported. Hepatic abnormalities are rare. Serious hematologic reactions have been noted, including thrombocytopenia and aplastic anemia. Hyperkalemia has been reported and is related to inhibition of the synthesis of prostaglandins in the kidney. Renal papillary necrosis has also been observed. Bolus injections decrease organ blood flow and impair urinary output, although continuous infusion does not. A number of interactions with other drugs have been reported (see Appendix II). As is true for other potent cyclooxygenase inhibitors also, use of indomethacin should be avoided in patients with nasal polyps or angioedema, in whom asthma may be precipitated.
Ketoprofen
Ketoprofen is a propionic acid derivative that inhibits both cyclooxygenase (nonselectively) and lipoxygenase. Its pharmacokinetic characteristics are given in Table 36–1. Concurrent administration of probenecid elevates ketoprofen levels and prolongs its plasma half-life.
The effectiveness of ketoprofen at dosages of 100–300 mg/d is equivalent to that of other NSAIDs in the treatment of rheumatoid arthritis, osteoarthritis, gout, dysmenorrhea, and other painful conditions. In spite of its dual effect on prostaglandins and leukotrienes, ketoprofen is not superior to other NSAIDs. Its major adverse effects are on the gastrointestinal tract and the central nervous system.
Ketorolac
Ketorolac is an NSAID promoted for systemic use mainly as an analgesic, not as an antiinflammatory drug (though it has typical NSAID properties). Pharmacokinetics are presented in Table 36–1. The drug does appear to have significant analgesic efficacy and has been used
successfully to replace morphine in some situations involving mild to moderate postsurgical pain. It is most often given intramuscularly or intravenously, but an oral dose formulation is available. When used with an opioid, it may decrease the opioid requirement by 25–50%. An ophthalmic preparation is available for anti-inflammatory applications. Toxicities are similar to those of other NSAIDs, although renal toxicity may be more common with chronic use.
Meclofenamate & Mefenamic Acid
Meclofenamate and mefenamic acid (Table 36–1) inhibit both COX and phospholipase A2. Meclofenamate appears to have adverse effects similar to those of other NSAIDs, though diarrhea and abdominal pain may be more common; it has no advantages over other NSAIDs. This drug enhances the effect of oral anticoagulants. Meclofenamate is contraindicated in pregnancy; its efficacy and safety have not been established for young children.
Mefenamic acid is probably less effective than aspirin as an anti-inflammatory agent and is clearly more toxic. It should not be used for longer than 1 week and should not be given to children.
Nabumetone
Nabumetone is the only nonacid NSAID in current use; it is converted to the active acetic acid derivative in the body. It is given as a ketone prodrug that resembles naproxen in structure (Figure 36–1). Its half-life of more than 24 hours (Table 36–1) permits once-daily dosing, and the drug does not appear to undergo enterohepatic circulation. Renal impairment results in a doubling of its halflife and a 30% increase in area under the curve. Its properties are very similar to those of other NSAIDs, though it may be less damaging to the stomach than some other NSAIDs when given at a dosage of 1000 mg/d. Unfortunately, higher doses (eg, 1500–2000 mg/d) are often needed, and this is a very expensive NSAID. Like naproxen, nabumetone has been reported to cause pseudoporphyria and photosensitivity in some patients. Other adverse effects mirror those of other NSAIDs.
Naproxen
Naproxen is a naphthylpropionic acid derivative. It is the only NSAID presently marketed as a single enantiomer, and it is a nonselective COX inhibitor. Naproxen's free fraction is 41% higher in women than in men, though albumin binding is very high in both sexes (Table 36–1). Naproxen is effective for the usual rheumatologic indications and is available both in a slow-release formulation and as an oral suspension. A topical preparation and an ophthalmic solution are also available.
The incidence of upper gastrointestinal bleeding in OTC use is low but still double that of OTC ibuprofen (perhaps due to a dose effect). Rare cases of allergic pneumonitis, leukocytoclastic vasculitis, and pseudoporphyria as well as the more common NSAID-associated adverse effects have been noted.
Oxaprozin
Oxaprozin is another propionic acid derivative NSAID. As noted in Table 36–1, its major difference from the other members of this subgroup is a very long half-life (50–60 hours), though oxaprozin does not undergo enterohepatic circulation. Because of its long half-life, oxaprozin can be given once a day, and dosage adjustments should be made at intervals no shorter than 5 days. The drug appears to have the same benefits and risks that are associated with other NSAIDs. It is mildly uricosuric, making it potentially more useful in gout than some other NSAIDs.
Phenylbutazone
Phenylbutazone, a pyrazolone derivative, rapidly gained favor after its introduction in 1949 for the treatment of rheumatic syndromes, but its toxicities—particularly the hematologic effects (including aplastic anemia)—have resulted in its withdrawal from the North American and most European markets. It is rarely used today.
Piroxicam
Piroxicam, an oxicam (Figure 36–1), is a nonselective COX inhibitor but at high concentrations also inhibits polymorphonuclear leukocyte migration, decreases oxygen radical production, and inhibits lymphocyte function. Its long half-life (Table 36–1) permits once-daily dosing.
Piroxicam can be used for the usual rheumatic indications. Toxicity includes gastrointestinal symptoms (20% of patients), dizziness, tinnitus, headache, and rash. When piroxicam is used in dosages higher than 20 mg/d, an increased incidence of peptic ulcer and bleeding is encountered. Epidemiologic studies suggest that this risk is as much as 9.5 times higher with piroxicam than with other NSAIDs.
Sulindac
Sulindac is a sulfoxide prodrug. It is reversibly metabolized to the active sulfide metabolite, which is excreted in bile and then reabsorbed from the intestine. The enterohepatic cycling prolongs the duration of action to 12–16 hours.
The indications and adverse reactions of sulindac are similar to those of other NSAIDs. In addition to its rheumatic disease indications, sulindac suppresses familial intestinal polyposis; it may inhibit the development of colon, breast, and prostate cancer in humans. It appears to inhibit the occurrence of gastrointestinal cancer in rats. The latter effect may be caused by the sulfone rather than the sulfide.
Because the sulfide may be reoxidized to the inactive prodrug in the kidney, sulindac may inhibit renal COX less than other NSAIDs, though reversible renal failure and nephrotic syndrome have been observed with this drug. Among the more severe reactions, Stevens-Johnson epidermal necrolysis syndrome, thrombocytopenia, agranulocytosis, and nephrotic syndrome have all been observed. Like diclofenac, sulindac may have some propensity to cause elevation of serum aminotransferases; it is also sometimes associated with cholestatic liver damage, which disappears or becomes quiescent when the drug is stopped.
Tenoxicam
Tenoxicam is an oxicam similar to piroxicam and shares its nonselective COX inhibition, long halflife (72 hours), efficacy, and toxicity profile. It is available abroad but not in the USA.
Tiaprofen
Tiaprofen is a racemic propionic acid derivative but does not undergo stereoconversion. It has a short serum half-life (1–2 hours) with an increase to 2–4 hours in the elderly. This drug inhibits renal uric acid reabsorption and thus decreases serum uric acid slightly. It is available for oral and intramuscular administration. Its efficacy and adverse event profiles mirror those of other NSAIDs, but tiaprofen is not available in the USA.

Tolmetin
Tolmetin is a nonselective COX inhibitor. Its short half-life means that it must be given frequently, and it is therefore not often used. It is similar to other NSAIDs in efficacy except in gout, in which it is ineffective (for unknown reasons). Its toxicity profile is also similar to those of other NSAIDs, with the rare additional problem of allergic IgM-related thrombocytopenic purpura.
Azapropazone & Carprofen
These drugs are available in many other countries but are not sold in the USA. Azapropazone (apazone), a pyrazolone derivative, is structurally related to phenylbutazone but appears less likely to cause agranulocytosis. Its half-life of 12–16 hours may be doubled in patients with decreased renal function. Carprofen is a propionic acid derivative with a half-life of 10–16 hours. The indications and adverse effects of azapropazone and carprofen are similar to those of other NSAIDs.
Clinical Pharmacology of the NSAIDs
All NSAIDs, including aspirin, are about equally efficacious with a few exceptions—tolmetin seems not to be effective for gout, and aspirin is less effective than other NSAIDs (eg, indomethacin) for ankylosing spondylitis. Thus, NSAIDs tend to be differentiated on the basis of toxicity and cost-effectiveness. For example, the gastrointestinal and renal side effects of ketorolac limit its use. Fries et al (1993), using a toxicity index, estimated that indomethacin, tolmetin, and meclofenamate were associated with the greatest toxicity, while salsalate, aspirin, and ibuprofen were least toxic. The selective COX-2 inhibitors were not included in this analysis.
For patients with renal insufficiency, nonacetylated salicylates may be best. Fenoprofen is less used because of its rare association with interstitial nephritis. Diclofenac and sulindac are associated with more liver function test abnormalities than other NSAIDs. The relatively expensive and selective COX-2 inhibitors are probably safest for patients at high risk for gastrointestinal bleeding. These drugs or a nonselective NSAID plus omeprazole or misoprostol may be appropriate in those patients at highest risk for gastrointestinal bleeding; in this subpopulation of patients, they are cost-effective despite their high acquisition costs.
The choice of an NSAID thus requires a balance of efficacy, cost-effectiveness, safety, and numerous personal factors (eg, other drugs also being used, concurrent illness, compliance, medical insurance coverage), so that there is no "best" NSAID for all patients. There may, however, be one or two best NSAIDs for a specific person.
Katzung PHARMACOLOGY, 9e > Section VI. Drugs Used to Treat Disease of the Blood, Inflammation, & Gout > Chapter 36. Nonsteroidal Anti-Inflammatory Drugs, Disease-Modifying Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout >
Disease-Modifying Antirheumatic Drugs (DMARDs)
Careful clinical and epidemiologic studies have shown that rheumatoid arthritis is an immunologic disease that causes significant systemic effects which shorten life in addition to the joint disease that reduces mobility and quality of life. NSAIDs offer mainly symptomatic relief; they reduce inflammation and the pain it causes and often preserve function, but they have little effect on the progression of bone and cartilage destruction. Interest has therefore centered on finding treatments that might arrest—or at least slow—this progression by modifying the disease itself. The effects of disease-modifying therapies may take 6 weeks to 6 months to become evident, ie, they are slow-
acting compared with NSAIDs. These therapies include methotrexate, azathioprine, penicillamine, hydroxychloroquine and chloroquine, organic gold compounds, sulfasalazine, leflunomide, tumor necrosis factor (TNF)-blocking agents, and immunoadsorption apheresis. Considerable controversy surrounds the long-term efficacy of many of these therapies.
Methotrexate
Methotrexate is now considered the first DMARD of choice in the treatment of rheumatoid arthritis and is used in up to 60% of patients. It is active in this condition at much lower doses than those needed in cancer chemotherapy (see Chapter 55: Cancer Chemotherapy).
Mechanism of Action
Methotrexate's principal mechanism of action at the low doses used in the rheumatic diseases probably relates to inhibition of aminoimidazolecarboxamide (AICAR) transformylase and thymidylate synthetase, with secondary effects on polymorphonuclear chemotaxis. While there is some effect on dihydrofolate reductase—and this effects lymphocyte and macrophage function—it is more likely its effect on AICAR transformylase that accounts for the major portion of its action in autoimmune disease.
Pharmacokinetics
The drug is approximately 70% absorbed after oral administration (see Chapter 55: Cancer Chemotherapy). It is metabolized to a less active hydroxylated metabolite, and both the parent compound and the metabolite are polyglutamated within cells, where they stay for prolonged periods. Methotrexate's serum half-life is usually only 6–9 hours, although it may be as long as 24 hours in some individuals. Methotrexate's concentration is increased in the presence of hydroxychloroquine. This drug is excreted principally in the urine, but up to 30% may be excreted in bile.
Indications
Although the most common methotrexate dosing regimens for the treatment of rheumatoid arthritis are 15 or 17.5 mg weekly, there is an increased effect up to 30 or 35 mg weekly. The drug decreases the rate of appearance of new erosions. Evidence supports its use in juvenile chronic arthritis, and it has been used in psoriasis, psoriatic arthritis, polymyositis, dermatomyositis, Wegener's granulomatosis, giant cell arteritis, subacute lupus erythematosus, and vasculitis.
Adverse Effects
Nausea and mucosal ulcers are the most common toxicities. Progressive dose-related hepatotoxicity in the form of enzyme elevation occurs frequently, but cirrhosis is rare (< 1%). Liver toxicity is not related to methotrexate concentrations, and liver biopsy follow-up is only recommended every 5 years. A rare "hypersensitivity" lung reaction with acute shortness of breath is documented, as are pseudolymphomatous reactions. The incidence of gastrointestinal and liver function test abnormalities can be reduced by the use of leucovorin 24 hours after each weekly dose or by the use of daily folic acid. This drug is contraindicated in pregnancy.
Chlorambucil
Mechanism of Action & Pharmacokinetics
Chlorambucil, probably through its metabolite phenylacetic acid mustard, cross-links DNA, thereby preventing cell replication. Its bioavailability is about 70% and it is completely metabolized, with excretion completed within 24 hours.
Indications
One controlled, double-blind trial plus anecdotal evidence attest to the efficacy of chlorambucil in rheumatoid arthritis. Chlorambucil has also been used in Beh_et's disease, systemic lupus erythematosus, vasculitis, and other autoimmune disorders.
Adverse Effects
The most common toxicity is dose-related bone marrow suppression. Infertility with azoospermia and amenorrhea also occurs. The risk of neoplasia is increased, with the relative risk of leukemia increased about tenfold compared with the general population, especially after more than 3 years of use.
Cyclophosphamide
Mechanism of Action
Cyclophosphamide's major active metabolite is phosphoramide mustard, which cross-links DNA to prevent cell replication. It suppresses T cell and B cell function by 30–40%; the T cell suppression correlates with clinical response in the rheumatic diseases.
Pharmacokinetics
See Chapter 55: Cancer Chemotherapy.
Indications
Cyclophosphamide is active against rheumatoid arthritis when given orally at dosages of 2 mg/kg/d but not when given intravenously. It is used regularly to treat systemic lupus erythematosus, vasculitis, Wegener's granulomatosis, and other severe rheumatic diseases.
Adverse Effects
Cyclophosphamide causes significant dose-related infertility in both men and women as well as bone marrow suppression, alopecia, hemorrhagic cystitis, and, rarely, bladder carcinoma (see Chapter 55: Cancer Chemotherapy).
Cyclosporine
Mechanism of Action
Through regulation of gene transcription, cyclosporine inhibits IL-1 and IL-2 receptor production and secondarily inhibits macrophage-T cell interaction and T cell responsiveness (see Chapter 56: Immunopharmacology). T cell-dependent B cell function is also affected.
Pharmacokinetics
Cyclosporine absorption is incomplete and somewhat erratic, although a new microemulsion formulation improves its consistency and provides 20 to 30% bioavailability. Grapefruit juice increases cyclosporine bioavailability up to 62%. Cyclosporine is metabolized by CYP3A and consequently is subject to a large number of drug interactions (see Chapter 56:
Immunopharmacology and Appendix II: Important Drug Interactions & Their Mechanisms).
Indications
Cyclosporine is approved for use in rheumatoid arthritis and retards the appearance of new bony erosions. Its usual dosage is 3–5 mg/kg/d divided into two doses. Anecdotal reports suggest that it may be useful in systemic lupus erythematosus, polymyositis and dermatomyositis, Wegener's granulomatosis, and juvenile chronic arthritis.
Adverse Effects
Cyclosporine has significant nephrotoxicity, and its toxicity can be increased by drug interactions with diltiazem, potassium-sparing diuretics, and other drugs inhibiting CYP3A. Serum creatinine should be closely monitored. Other toxicities include hypertension, hyperkalemia, hepatotoxicity, gingival hyperplasia, and hirsutism.
Azathioprine
Mechanism of Action
Azathioprine acts through its major metabolite, 6-thioguanine. 6-Thioguanine suppresses inosinic acid synthesis, B cell and T cell function, immunoglobulin production, and IL-2 secretion (see Chapter 56: Immunopharmacology).
Pharmacokinetics
The metabolism of azathioprine is bimodal, with rapid metabolizers clearing the drug four times more rapidly than slow metabolizers. Production of 6-thioguanine is dependent on thiopurine methyltransferase (TPMT), and patients with low or absent TPMT activity (0.3% of the population) are at particularly high risk of myelosuppression by excess concentrations of the parent drug if dosage is not adjusted.
Indications
Azathioprine is approved for use in rheumatoid arthritis and is used at a dosage of 2 mg/kg/d. Controlled trials show efficacy in psoriatic arthritis, reactive arthritis, polymyositis, systemic lupus erythematosus, and Behçet's disease
Adverse Effects
Azathioprine's toxicity includes bone marrow suppression, gastrointestinal disturbances, and some increase in infection risk. As noted in Chapter 56: Immunopharmacology, lymphomas may be increased with azathioprine use. Rarely, fever, rash, and hepatotoxicity signal acute allergic reactions.
Mycophenolate Mofetil
Mechanism of Action
Mycophenolate mofetil (MMF) is converted to myco-phenolic acid, the active form of the drug. The active product inhibits cytosine monophosphate dehydrogenase and secondarily, inhibits T cell lymphocyte proliferation; downstream, it interferes with leukocyte adhesion to endothelial cells through inhibition of E-selectin, P-selectin, and intercellular adhesion molecule 1.
Pharmacokinetics
See Chapter 56: Immunopharmacology.
Indications
MMF has been shown to be effective for the treatment of renal disease due to systemic lupus erythematosus and may be useful in vasculitis and Wegener's granulomatosis. While occasionally used at a dosage of 2 g/d to treat rheumatoid arthritis, there are few controlled data regarding its efficacy in this disease.
Adverse Effects
Comparisons with azathioprine in the renal transplantation literature show that MMF and azathioprine have similar gastrointestinal, hematopoietic, and hepatic toxicity profiles, with a possibly decreased incidence of fungal infections among patients treated with MMF. Hepatic toxicities are infrequent but must be monitored.
Chloroquine & Hydroxychloroquine
Mechanism of Action
Chloroquine and hydroxychloroquine are used mainly in malaria (see Chapter 53: Antiprotozoal Drugs). The mechanism of the anti-inflammatory action of these drugs in rheumatic diseases is unclear. The following mechanisms have been proposed: suppression of T lymphocyte responses to mitogens, decreased leukocyte chemotaxis, stabilization of lysosomal enzymes, inhibition of DNA and RNA synthesis, and the trapping of free radicals.
Pharmacokinetics
Antimalarials are rapidly absorbed but only 50% protein-bound in the plasma. They are very extensively tissue-bound, particularly in melanin-containing tissues such as the eyes. The drugs are deaminated in the liver and have blood elimination half-lives of up to 45 days.
Indications
Antimalarials are approved for rheumatoid arthritis, but they are not considered very efficacious DMARDs. Dose-response and serum concentration-response relationships have been documented for hydroxychloroquine. While antimalarials improve symptoms, there is no evidence that these compounds alter bony damage in rheumatoid arthritis at their usual dosages (up to 6.4 mg/kg/d hydroxychloroquine or 200 mg/d chloroquine). It usually takes 3–6 months to obtain a response. Antimalarials are often used for the treatment of the skin manifestations, serositis, and joint pains of

systemic lupus erythematosus, and they have been used in Sjögren's syndrome.
Adverse Effects
Although ocular toxicity may occur at dosages greater than 250 mg/d chloroquine and greater than 6.4 mg/kg/d hydroxychloroquine, it rarely occurs at lower doses. Nevertheless, ophthalmologic monitoring every 6–12 months is advised. Other toxicities include dyspepsia, nausea, vomiting, abdominal pain, rashes, and nightmares. These drugs appear to be relatively safe in pregnancy.
Gold
Gold compounds were first proved to be effective in a large double-blind trial in 1960. Because of their toxicity, they are used infrequently today. Their intramuscular formulations (aurothiomalate and aurothioglucose) contain 50% elemental gold. The oral formulation (auranofin) contains 29% elemental gold.
Mechanism of Action
Gold alters the morphology and functional capabilities of human macrophages—possibly its major mode of action. As a result, monocyte chemotactic factor-1, interleukin-8, interleukin-1 production, and vascular endothelial growth factor are all inhibited. Intramuscular gold compounds also alter lysosomal enzyme activity, reduce histamine release from mast cells, inactivate the first component of complement, and suppress the phagocytic activities of polymorphonuclear leukocytes. Auranofin also inhibits release of prostaglandin E2 and leukotriene B4.
production, and vascular endothelial growth factor are all inhibited. Intramuscular gold compounds also alter lysosomal enzyme activity, reduce histamine release from mast cells, inactivate the first component of complement, and suppress the phagocytic activities of polymorphonuclear leukocytes. Auranofin also inhibits release of prostaglandin E2 and leukotriene B4.
Pharmacokinetics
These compounds have high bioavailability after intramuscular administration and tend to concentrate in synovial membranes, liver, kidney, spleen, lymph nodes, and bone marrow. One month after an intramuscular injection, 75–80% of the drug is eliminated from the serum, but intramuscular gold's total body half-life is approximately 1 year. Auranofin is only about 25% bioavailable. Gold compounds are excreted approximately 66% in the urine and 33% via the feces. There has generally been no correlation found between serum gold concentration and either efficacy or toxicity.
Indications
Gold is effective for active rheumatoid arthritis and has been shown to slow radiologic progression of the disease. It has also been used in Sjögren's syndrome and juvenile rheumatoid arthritis, while use in psoriatic arthritis is controversial. In Japan, gold is used to treat asthma. The oral form of gold is effective in rheumatoid arthritis, but it appears less effective than the intramuscular formulation and is generally felt to have only modest effects.
Clinical Use
Intramuscular gold is given as a test dose of 5–25 mg and then as 50 mg intramuscular doses weekly for 20 weeks. Continued treatment, with maintained response, frequently allows lengthening of the dosing interval to 2, 3, or 4 weeks. Oral gold is generally given as 6 mg doses daily.
Adverse Effects
Pruritic skin rashes occur in 15–20% of patients, sometimes associated with eosinophilia. Stomatitis and a metallic taste in the mouth are common. Hematologic abnormalities, including thrombocytopenia, leukopenia, and even pancytopenia occur in 1–10% of patients. Aplastic anemia, while very rare, may be fatal. Eight to 10 percent of patients develop proteinuria that may progress to nephrotic syndrome. Other rare toxicities include enterocolitis, cholestatic jaundice, peripheral neuropathy, and pulmonary infiltrates. Corneal deposition of gold occurs but has little clinical import. Nitritoid reactions (sweating, flushing, and headaches) can occur, especially with gold thiomalate, and are presumably due to the vehicle rather than the gold salts. Adverse effects cause 30–40% of patients to discontinue gold therapy within a year.
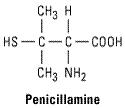
Penicillamine
Penicillamine, a metabolite of penicillin, is an analog of amino acid cystine. The D isomer has been used in rheumatoid arthritis. Penicillamine is rarely used today because of toxicity.
Sulfasalazine
Mechanism of Action
Sulfasalazine is metabolized to sulfapyridine and 5-aminosalicylic acid, and it is thought that the sulfapyridine is probably the active moiety when treating rheumatoid arthritis (unlike inflammatory bowel disease; see Chapter 63: Drugs Used in the Treatment of Gastrointestinal Diseases). Some authorities believe that the parent compound, sulfasalazine, also has an effect. In treated arthritis patients, IgA and IgM rheumatoid factor production are decreased. Suppression of T cell responses to concanavalin and inhibition of in vitro B cell proliferation have also been documented. It is not clear how these findings relate to the clinical efficacy of sulfasalazine in rheumatoid arthritis.
Pharmacokinetics
Only 10–20% of orally administered sulfasalazine is absorbed, although a fraction undergoes enterohepatic recirculation into the bowel, where sulfasalazine is reduced by intestinal bacteria to liberate sulfapyridine and 5-aminosalicylic acid. Sulfapyridine is well absorbed while 5- aminosalicylic acid remains unabsorbed. Some sulfasalazine is excreted unchanged in the urine whereas sulfapyridine is excreted after hepatic acetylation and hydroxylation. Sulfasalazine's halflife is 6–17 hours.
Indications
Sulfasalazine is effective in rheumatoid arthritis and reduces the rate of appearance of new joint damage. It has been used in juvenile chronic arthritis and ankylosing spondylitis and its associated uveitis. The usual regimen is 2–3 g/d.
Adverse Effects