

Книги фарма 2 / Bertram G. Katzung-Basic & Clinical Pharmacology(9th Edition)
.pdfIIb/IIIa, which binds fibrinogen and other macromolecules. Antiplatelet prostacyclin (PGI2) is released from the endothelium. Aggregating substances released from the degranulating platelet include ADP, TXA2, and 5-HT. Production of factor Xa is detailed in Figure 34–2. (Redrawn and reproduced, with permission, from Simoons ML, Decker JW: New directions in anticoagulant and antiplatelet treatment. [Editorial.] Br Heart J 1995;74:337.)
The platelet is central to normal hemostasis and to all thromboembolic disease. A white thrombus forms initially in high-pressure arteries by adherence of circulating platelets to areas of abnormal endothelium as described above. The growing thrombus of aggregated platelets reduces arterial flow. This localized stasis triggers fibrin formation, and a red thrombus forms around the nidal white thrombus.
A red thrombus can form around a white thrombus as mentioned above or de novo in low-pressure veins, initially by adherence of platelets (as in arteries) but followed promptly by the process of blood coagulation so that the bulk of the thrombus forms a long tail consisting of a fibrin network in which red cells are enmeshed. These tails become detached easily and travel as emboli to the pulmonary arteries. Such emboli often arise from a deep venous thrombosis (DVT)—a thrombus in the veins of the legs or pelvis. Although all thrombi are mixed, the platelet nidus dominates the arterial thrombus and the fibrin tail the venous thrombus. Arterial thrombi cause serious disease by producing local occlusive ischemia; venous thrombi, by giving rise to distant embolization.
Blood Coagulation
Blood coagulates by the transformation of soluble fibrinogen into insoluble fibrin. Several circulating proteins interact in a cascading series of limited proteolytic reactions. At each step, a clotting factor zymogen (eg, factor VII) undergoes limited proteolysis and becomes an active protease (eg, factor VIIa). Thus, each protease factor activates the next clotting factor until finally a solid fibrin clot is formed. Fibrinogen (factor I), the soluble precursor of fibrin, is the substrate for the enzyme thrombin (factor IIa). This protease is formed during coagulation by activation of its zymogen, prothrombin (factor II). Prothrombin is bound by calcium to a platelet phospholipid (PL) surface, where activated factor X (Xa), in the presence of factor Va, converts it into circulating thrombin. Several of the blood clotting factors are targets for drug therapy (Table 34–1).
Table 34–1. Blood Clotting Factors and Drugs That Affect Them.1
Component or |
Common Synonym |
Target for the Action of: |
Factor |
|
|
|
|
|
I |
Fibrinogen |
|
|
|
|
II |
Prothrombin |
Heparin (IIa); warfarin |
|
|
(synthesis) |
|
|
|
III |
Tissue thromboplastin |
|
|
|
|
IV |
Calcium |
|
|
|
|
V |
Proaccelerin |
|
|
|
|
VII |
Proconvertin |
Warfarin (synthesis) |
|
|
|
VIII |
Antihemophilic factor (AHF) |
|
|
|
|
IX |
Christmas factor, plasma thromboplastin |
Warfarin (synthesis) |
|
|
|
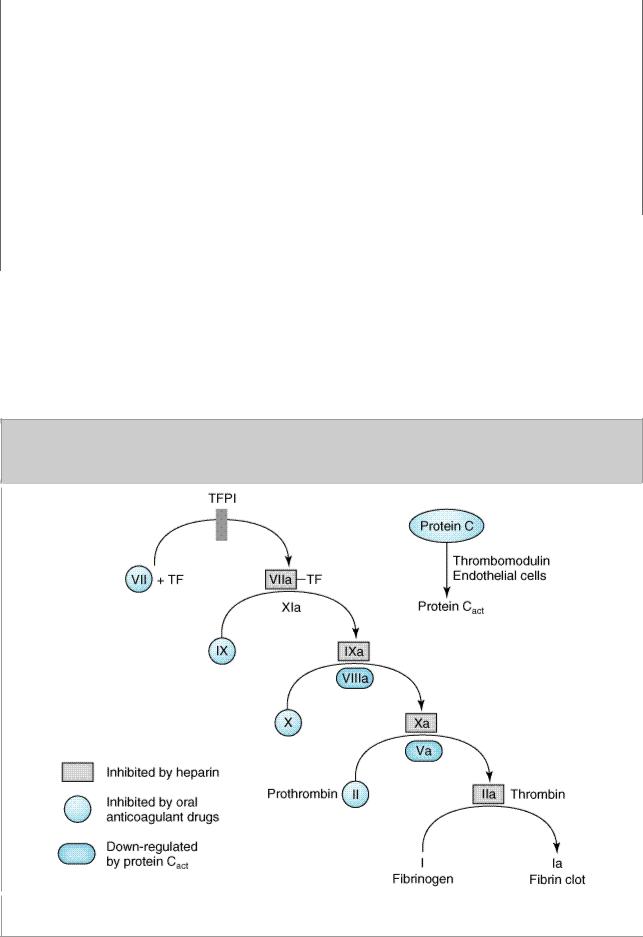

catalyzes this reaction. Tissue factor pathway inhibitor (TFPI) inhibits the catalytic action of the VIIa-TF complex. The cascade proceeds as shown, resulting ultimately in the conversion of fibrinogen to fibrin, an essential component of a functional clot. The two major anticoagulant drugs, heparin and warfarin (an oral anticoagulant), have very different actions. Heparin, acting in the blood, directly activates anticlotting factors, specifically antithrombin, which inactivates the factors enclosed in rectangles. Warfarin, acting in the liver, inhibits the synthesis of the factors enclosed in circles. Proteins C and S exert anticlotting effects by inactivating activated factors Va and VIIIa.
The TF/factor VII/factor X process is inhibited and regulated by tissue factor pathway inhibitor (TFPI). Oral anticoagulant drugs inhibit the hepatic synthesis of several clotting factors. Heparin inhibits the activity of several of these activated clotting factors by enhancing the anticoagulant activity of antithrombin, which inactivates the serine proteases IIa, IXa, Xa, XIa, and XIIa. The endogenous anticoagulants protein C and protein S diminish amplification in the blood clotting cascade by proteolysis of factors Va and VIIIa.
Regulation of Coagulation & Fibrinolysis
Blood coagulation and thrombus formation must be confined to the smallest possible area to achieve local hemostasis in response to bleeding from trauma or surgery without causing disseminated coagulation or impaired blood flow. Two major systems regulate and delineate these processes: fibrin inhibition and fibrinolysis.
Plasma contains protease inhibitors that rapidly inactivate the coagulation proteins as they escape from the site of vessel injury. The most important proteins of this system are 
 1-antiprotease,
1-antiprotease, 
 2- macroglobulin,
2- macroglobulin, 
 2-antiplasmin, and antithrombin. If this system is overwhelmed, generalized intravascular clotting may occur. This process is called disseminated intravascular coagulation (DIC) and may follow massive tissue injury, cell lysis in malignant neoplastic disease, obstetric emergencies such as abruptio placentae, or bacterial sepsis.
2-antiplasmin, and antithrombin. If this system is overwhelmed, generalized intravascular clotting may occur. This process is called disseminated intravascular coagulation (DIC) and may follow massive tissue injury, cell lysis in malignant neoplastic disease, obstetric emergencies such as abruptio placentae, or bacterial sepsis.
The central process of fibrinolysis is conversion of inactive plasminogen to the proteolytic enzyme plasmin. Injured cells release activators of plasminogen. Plasmin remodels the thrombus and limits the extension of thrombosis by proteolytic digestion of fibrin.
Regulation of the fibrinolytic system is useful in therapeutics. Increased fibrinolysis is effective therapy for thrombotic disease. Tissue plasminogen activator (t-PA), urokinase, and streptokinase all activate the fibrinolytic system (Figure 34–3). Conversely, decreased fibrinolysis protects clots from lysis and reduces the bleeding of hemostatic failure. Aminocaproic acid is a clinically useful inhibitor of fibrinolysis. Heparin and the oral anticoagulant drugs do not affect the fibrin-olytic mechanism.
Figure 34–3.

Schematic representation of the fibrin-olytic system. Plasmin is the active fibrinolytic enzyme. Several clinically useful activators are shown on the left in bold. Anistreplase is a combination of streptokinase and the proactivator plasminogen. Aminocaproic acid (right) inhibits the activation of plasminogen to plasmin and is useful in some bleeding disorders.
Katzung PHARMACOLOGY, 9e > Section VI. Drugs Used to Treat Disease of the Blood, Inflammation, & Gout > Chapter 34. Drugs Used in Disorders of Coagulation >
Basic Pharmacology of the Anticoagulant Drugs
Indirect Thrombin Inhibitors
The indirect thrombin inhibitors are so named because their antithrombotic effect is exerted by their interaction with antithrombin. Unfractionated heparin (UFH), low-molecular-weight heparin (LMWH), and the synthetic pentasaccharide fondaparinux bind to antithrombin and enhance its inactivation of factor Xa. UFH and to a lesser extent LMWH also enhance antithrombin's inactivation of thrombin (IIa).
Heparin
Chemistry & Mechanism of Action
Heparin is a heterogeneous mixture of sulfated mucopolysaccharides. It binds to endothelial cell surfaces and a variety of plasma proteins. As noted above, its biologic activity is dependent upon the plasma protease inhibitor antithrombin. Antithrombin inhibits clotting factor proteases, especially thrombin (IIa), IXa, and Xa, by forming equimolar stable complexes with them. In the absence of heparin, these reactions are slow; in the presence of heparin, they are accelerated 1000fold. Only about a third of the molecules in commercial heparin preparations have an accelerating effect because the remainder lack the unique pentasaccharide sequence needed for high-affinity binding to antithrombin (Figure 34–4). The active heparin molecules bind tightly to antithrombin

and cause a conformational change in this inhibitor. The conformational change of antithrombin exposes its active site for more rapid interaction with the proteases (the activated clotting factors). Heparin catalyzes the antithrombin-protease reaction without being consumed. Once the antithrombin-protease complex is formed, heparin is released intact for renewed binding to more antithrombin.
Figure 34–4.
Subunit structure of heparin. The small polymer section shown illustrates the repeating disaccharide units typical of heparin. The sequence shows the critical pentasaccharide portion required for binding to antithrombin. In addition to those shown, other saccharides occur. Heparin is a strongly acidic molecule because of its high content of anionic sulfate and carboxylic acid groups. GlcN, glucosamine; IdUA, iduronic acid; GlcUA, glucuronic acid. The same five residues with the terminal groups shown in parentheses, constitute fondaparinux.
The antithrombin binding region of commercial unfractionated heparin consists of repeating sulfated disaccharide units composed of D-glucosamine-L-iduronic acid and D-glucosamine-D- glucuronic acid (Figure 34–4). High-molecular-weight (HMW) fractions of heparin with high affinity for antithrombin markedly inhibit blood coagulation by inhibiting all three factors, especially thrombin and factor Xa. Unfractionated heparin has a MW range of 5000–30,000. In contrast, the shorter-chain low-molecular-weight (LMW) fractions of heparin inhibit activated factor X but have less effect on thrombin (and on coagulation in general) than the HMW species. Nevertheless, numerous studies have demonstrated that LMW heparins such as enoxaparin, dalteparin, and tinzaparin are effective in several thromboembolic conditions. In fact, these LMW heparins—in comparison with UFH—have equal efficacy, increased bioavailability from the subcutaneous site of injection, and less frequent dosing requirements (once or twice daily is sufficient).
Because commercial heparin consists of a family of molecules of different molecular weights, the correlation between the concentration of a given heparin preparation and its effect on coagulation often is poor. Therefore, UFH is standardized by bioassay. Heparin sodium USP must contain at least 120 USP units per milligram. Heparin is generally used as the sodium salt, but calcium heparin is equally effective. Lithium heparin is used in vitro as an anticoagulant for blood samples. Commercial heparin is extracted from porcine intestinal mucosa and bovine lung. Enoxaparin is obtained from the same sources as regular heparin, but doses are specified in milligrams. Dalteparin, tinzaparin and danaparoid (an LMW heparanoid containing heparan sulfate, dermatan sulfate, and chondroitin sulfate that is no longer available in the United States), on the other hand, are specified in anti-factor Xa units.
Toxicity
The major adverse effect of heparin is bleeding. This risk can be decreased by scrupulous patient selection, careful control of dosage, and close monitoring of the activated partial thromboplastin time (aPTT) in those patients receiving unfractionated heparin. Levels for UFH may also be determined by protamine titration (therapeutic levels 0.2–0.4 unit/mL) or anti-Xa units (therapeutic levels 0.3–0.7 unit/mL). Weight-based dosing of the LMW heparins results in predictable pharmacokinetics and plasma levels in patients with normal renal function. Therefore, LMW heparin levels are not generally measured except in the setting of renal insufficiency, obesity, and pregnancy. LMW heparin levels are determined by anti-Xa units. Peak therapeutic levels are 0.5–1 unit/mL for twice daily dosing, determined 4 hours after administration, and approximately 1.5 units/mL for once daily dosing. Elderly women and patients with renal failure are more prone to hemorrhage. Heparin is of animal origin and should be used cautiously in patients with allergy. Increased loss of hair and reversible alopecia have been reported. Long-term heparin therapy is associated with osteoporosis and spontaneous fractures. Heparin accelerates the clearing of postprandial lipemia by causing the release of lipoprotein lipase from tissues, and long-term use is associated with mineralocorticoid deficiency.
Heparin causes transient thrombocytopenia in 25% or more of patients and severe thrombocytopenia in 5%. Mild platelet reduction within the first 5 days of therapy may result from heparin-induced aggregation that is postulated to be benign and transient in character. A smaller subset of patients may develop an antibody-mediated thrombocytopenia that is associated with paradoxical thrombosis. In these instances, the heparin-induced antibody is directed against the heparin-platelet factor 4 complex. These antigen-antibody complexes bind to Fc receptors on adjacent platelets, causing aggregation and thromboembolism. The following points should be considered in all patients receiving heparin: Platelet counts should be performed frequently; thrombocytopenia should be considered to be heparin-induced; any new thrombus can be the result of heparin; and thromboembolic disease thought to be heparin-induced should be treated by discontinuance of heparin and administration of an alternative drug, such as a direct thrombin inhibitor (see below). Administration of warfarin alone is contraindicated since it may exacerbate the prothrombotic state associated with heparin-induced thrombocytopenia.
Contraindications
Heparin is contraindicated in patients who are hypersensitive to the drug, are actively bleeding, or have hemophilia, significant thrombocytopenia, purpura, severe hypertension, intracranial hemorrhage, infective endocarditis, active tuberculosis, ulcerative lesions of the gastrointestinal tract, threatened abortion, visceral carcinoma, or advanced hepatic or renal disease. Heparin should be avoided in those patients who have recently had surgery of the brain, spinal cord, or eye and in patients who are undergoing lumbar puncture or regional anesthetic block. Despite the apparent lack of placental transfer, heparin should be used in pregnant women only when clearly indicated.
Administration & Dosage
The indications for the use of heparin are described in the section on clinical pharmacology. A plasma concentration of heparin of 0.2–0.4 unit/mL (by protamine titration) or 0.3–0.7 unit/mL (anti-Xa units) usually prevents pulmonary emboli in patients with established venous thrombosis. This concentration of heparin will prolong the activated partial thromboplastin time (aPTT) to 2–2.5 times that of the control value. This degree of anticoagulant effect should be maintained throughout the course of continuous intravenous heparin therapy. When intermittent heparin administration is used, the aPTT should be measured 6 hours after the administered dose to maintain prolongation of the aPTT to 2–2.5 times that of the control value.
Continuous intravenous administration of heparin is accomplished via an infusion pump. After an initial bolus injection of 80-100 units/kg, a continuous infusion of about 15-22 units/kg/h is required to maintain the aPTT at 2–2.5 times control. Patients with acute pulmonary emboli often require larger doses than these during the first few days because of binding to a variety of acute phase proteins, such as factor VIII and von Willebrand factor, and increased heparin clearance. Subcutaneous administration of heparin, as in low-dose prophylaxis, is achieved with 5000 units every 8–12 hours. Because of the danger of hematoma formation at the injection site, heparin must never be administered intramuscularly.
Prophylactic enoxaparin is given subcutaneously in a dosage of 30 mg twice daily or 40 mg once daily. Full-dose enoxaparin therapy is 1 mg/kg subcutaneously every 12 hours. This corresponds to a therapeutic anti-factor Xa level of 0.5–1 unit/mL. Selected patients may be treated with enoxaparin 1.5 mg/kg once a day, with a target anti-Xa level of 1.5 units/mL. The prophylactic dose of dalteparin is 5000 units subcutaneously once a day; therapeutic dosing is 200 units/kg once a day for venous disease or 120 units/kg every 12 hours for acute coronary syndrome. The use of LMW heparins is discouraged or contraindicated in patients with renal insufficiency or body weight greater than 150 kg.
The synthetic pentasaccharide molecule fondaparinux (Figure 34–4) avidly binds antithrombin with high specific activity, resulting in efficient inactivation of factor Xa. Fondaparinux has a long halflife of 15 hours, allowing for once-daily dosing by subcutaneous administration. A series of phase 3 studies in orthopedic patients comparing fondaparinux 2.5 mg subcutaneously daily beginning 6 hours postoperatively versus enoxaparin either 40 mg daily or 30 mg twice daily found fondaparinux to be superior in preventing development of asymptomatic deep venous thrombosis. A phase 3 study evaluating the use of fondaparinux coupled with warfarin compared with LMWH plus warfarin in the treatment of acute venous thromboembolic disease found these two treatment approaches equivalent. Increased bleeding with fondaparinux is seen in patients administered the drug sooner than 6 hours postoperatively, in those who weigh less than 50 kg, and in those with renal insufficiency.
Reversal of Heparin Action
Excessive anticoagulant action of heparin is treated by discontinuance of the drug. If bleeding occurs, administration of a specific antagonist such as protamine sulfate is indicated. Protamine is a highly basic peptide that combines with heparin as an ion pair to form a stable complex devoid of anticoagulant activity. For every 100 units of heparin remaining in the patient, administer 1 mg of protamine sulfate intravenously; the rate of infusion should not exceed 50 mg in any 10-minute period. Excess protamine must be avoided; it also has an anticoagulant effect. Neutralization of LMW heparin by protamine is incomplete. Limited experience suggests that 1 mg of protamine sulfate may be used to partially neutralize 1 mg of enoxaparin. Protamine will not reverse the activity of fondaparinux. Excess danaparoid can be removed by plasmapheresis.
Direct Thrombin Inhibitors
The direct thrombin inhibitors (DTI) are a relatively new class of agents that exert their anticoagulant effect by directly binding to the active site of thrombin, thereby inhibiting thrombin's downstream effects. The DTIs bind thrombin without additional binding proteins, such as antithrombin, and they do not bind to other plasma proteins, such as platelet factor 4. Hirudin and bivalirudin are bivalent DTIs in that they bind at both the catalytic or active site of thrombin as well as at a substrate recognition site. Argatroban and melagatran are small molecules that bind only at the thrombin active site.

Hirudin
For a number of years, surgeons have used medicinal leeches (Hirudo medicinalis) to prevent thrombosis in the fine vessels of reattached digits. Hirudin is a specific, irreversible thrombin inhibitor from the leech that is now available in recombinant form as lepirudin. Its action is independent of antithrombin, which means it can reach and inactivate fibrin-bound thrombin in thrombi. Lepirudin has little effect on platelets or the bleeding time. Like heparin, it must be administered parenterally and is monitored by the aPTT. Lepirudin is FDA-approved for use in patients with thrombosis related to heparin-induced thrombocytopenia. This drug has a short halflife, but it accumulates in renal insufficiency and no antidote exists. Up to 40% of patients on longterm infusions develop an antibody directed against the thrombin-lepirudin complex. These antigenantibody complexes are not cleared by the kidney and may result in an enhanced anticoagulant effect.
Bivalirudin, another bivalent inhibitor of thrombin, is administered intravenously, with a rapid onset and offset of action. The drug has a short half-life with clearance that is 20% renal and the remainder metabolic. Bivalirudin inhibits platelet activation and been FDA-approved for use in percutaneous coronary angioplasty.
Argatroban is a small molecule thrombin inhibitor that is FDA approved for use in patients with heparin-induced thrombocytopenia (HIT) with or without thrombosis and coronary angioplasty in patients with HIT. It, too, has a short half-life, is given by continuous intravenous infusion, and monitoring is done by aPTT. Its clearance is not affected by renal disease but is dependent on liver function. The drug requires dose reduction in patients with liver disease. Patients on argatroban will demonstrate elevated INRs because of test interference, rendering the transition to warfarin difficult.
Melagatran is the fourth parenteral drug in this class. It and its oral prodrug, ximelagatran, are under intensive study. Attractive features of ximelagatran include predictable pharmacokinetics and bioavailability—allowing for fixed dosing and predictable anticoagulant response; no need for routine coagulation monitoring; lack of interaction with P450-interacting drugs; rapid onset and offset of action—allowing for immediate anticoagulation and thus no need for overlap with additional anticoagulant drugs. A published phase 3 trial in patients status post major orthopedic surgery found ximelagatran equivalent to warfarin in preventing postoperative DVT. Clinical trials in patients with acute DVT and chronic atrial fibrillation are on-going.
Warfarin & the Coumarin Anticoagulants
Chemistry & Pharmacokinetics
The clinical use of the coumarin anticoagulants can be traced to the discovery of an anticoagulant substance formed in spoiled sweet clover silage. It produced a deficiency of plasma prothrombin and consequent hemorrhagic disease in cattle. The toxic agent was identified as bishydroxycoumarin and synthesized as dicumarol. This drug and its congeners, most notably warfarin (Figure 34–5), are widely used as rodenticides in addition to their application as antithrombotic agents in humans. Warfarin is the most reliable member of this group, and the other coumarin anticoagulants are almost never used in the USA.
Figure 34–5.

Structural formulas of several oral anticoagulant drugs and of vitamin K. The carbon atom of warfarin shown at the asterisk is an asymmetric center.
Warfarin is generally administered as the sodium salt and has 100% bioavailability. Over 99% of racemic warfarin is bound to plasma albumin, which may contribute to its small volume of distribution (the albumin space), its long half-life in plasma (36 hours), and the lack of urinary excretion of unchanged drug. Warfarin used clinically is a racemic mixture composed of equal amounts of two enantiomorphs. The levorotatory S-warfarin is four times more potent than the dextrorotatory R-warfarin. This observation is useful in understanding the stereoselective nature of several drug interactions involving warfarin.
Mechanism of Action
Coumarin anticoagulants block the  -carboxylation of several glutamate residues in prothrombin and factors VII, IX, and X as well as the endogenous anticoagulant proteins C and S (Figure 34–2). The blockade results in incomplete molecules that are biologically inactive in coagulation. This protein carboxylation is physiologically coupled with the oxidative deactivation of vitamin K. The anticoagulant prevents reductive metabolism of the inactive vitamin K epoxide back to its active hydroquinone form (Figure 34–6). Mutational change of the responsible enzyme, vitamin K epoxide reductase, can give rise to genetic resistance to warfarin in humans and especially in rats.
-carboxylation of several glutamate residues in prothrombin and factors VII, IX, and X as well as the endogenous anticoagulant proteins C and S (Figure 34–2). The blockade results in incomplete molecules that are biologically inactive in coagulation. This protein carboxylation is physiologically coupled with the oxidative deactivation of vitamin K. The anticoagulant prevents reductive metabolism of the inactive vitamin K epoxide back to its active hydroquinone form (Figure 34–6). Mutational change of the responsible enzyme, vitamin K epoxide reductase, can give rise to genetic resistance to warfarin in humans and especially in rats.
Figure 34–6.

Vitamin K cycle—metabolic interconversions of vitamin K associated with the synthesis of vitamin K-dependent clotting factors. Vitamin K1 or K2 is activated by reduction to the hydroquinone form (KH2). Stepwise oxidation to vitamin K epoxide (KO) is coupled to prothrombin carboxylation by the enzyme carboxylase. The reactivation of vitamin K epoxide is the warfarin-sensitive step (warfarin). The R on the vitamin K molecule represents a 20-carbon phytyl side chain in vitamin K1 and a 30to 65-carbon polyprenyl side chain in vitamin K2.
There is an 8- to 12-hour delay in the action of warfarin. Its anticoagulant effect results from a balance between partially inhibited synthesis and unaltered degradation of the four vitamin K- dependent clotting factors. The resulting inhibition of coagulation is dependent on their degradation rate in the circulation. These half-lives are 6, 24, 40, and 60 hours for factors VII, IX, X, and II, respectively. Larger initial doses of warfarin—up to about 0.75 mg/kg—hasten the onset of the anticoagulant effect. Beyond this dosage, the speed of onset is independent of the dose size. The only effect of a larger loading dose is to prolong the time that the plasma concentration of drug remains above that required for suppression of clotting factor synthesis. The only difference among oral anticoagulants in producing and maintaining hypoprothrombinemia is the half-life of each drug.
Toxicity
Warfarin crosses the placenta readily and can cause a hemorrhagic disorder in the fetus. Furthermore, fetal proteins with  -carboxyglutamate residues found in bone and blood may be affected by warfarin; the drug can cause a serious birth defect characterized by abnormal bone formation. Thus, warfarin should never be administered during pregnancy. Cutaneous necrosis with reduced activity of protein C sometimes occurs during the first weeks of therapy. Rarely, the same process causes frank infarction of breast, fatty tissues, intestine, and extremities. The pathologic lesion associated with the hemorrhagic infarction is venous thrombosis, suggesting that it is caused by warfarin-induced depression of protein C synthesis.
-carboxyglutamate residues found in bone and blood may be affected by warfarin; the drug can cause a serious birth defect characterized by abnormal bone formation. Thus, warfarin should never be administered during pregnancy. Cutaneous necrosis with reduced activity of protein C sometimes occurs during the first weeks of therapy. Rarely, the same process causes frank infarction of breast, fatty tissues, intestine, and extremities. The pathologic lesion associated with the hemorrhagic infarction is venous thrombosis, suggesting that it is caused by warfarin-induced depression of protein C synthesis.