

Reactive Intermediate Chemistry
.pdf214 ORGANIC RADICAL IONS
post-Columbian accounts of Eldorado, all based on ‘‘direct’’ observation. Alas, they represent gross misinterpretations.
Frequently, the general nature or detailed structure of an intermediate is inferred from reaction products. Radical ions are invoked in reactions between electron donors and acceptors in polar solvents. Probing the fate of chirality, stereochemistry, or an isotopic label or substituent in the products of a chemical transformation exemplifies the classical approach to mechanism. Of course, this approach is not without shortcomings.
4. RADICAL ION STRUCTURES
Because radical ions are formed by removing an electron from the HOMO or adding an electron to the LUMO of a substrate, their singly occupied molecular orbitals (SOMOs) and their structures are determined to some extent by the nature of the HOMO or LUMO of the parent molecule. The relationship between parent and radical ion structure has been probed particularly for cations. Electron ejection in the gas phase (in PES spectroscopy) proceeds vertically, generating radical cations with the geometry of the parent. Some cations maintain structures closely related to the neutral parent even on longer time scales; others relax to lower-energy structures with changes in bond lengths and bond and dihedral angles. Particularly strained ring compounds may undergo major structural changes upon ionization. In the extreme case, a bond may break, giving rise to radical ions whose spin and charge are localized on two different atoms (or sections) of the molecule; such species are called distonic radical ions. Possible relationships between the geometries of radical cations and their precursors include the purely vertical relationship, minor changes (relaxation), or major changes in geometry (rearrangement, Fig. 6.6).
Depending on the HOMO or LUMO involved in the redox reaction, we differentiate between p, n, or s donors and p or s acceptors, respectively; in addition,
Figure 6.6. Schematic comparison between potential surfaces of radical cations and their precursors. (a) Vertical ionization without relaxation. (b) Vertical ionization followed by relaxation. (c) Vertical ionization followed by rearrangement.
RADICAL ION STRUCTURES |
215 |
some substrates contain a combination of functions. These families show different degrees of structural changes upon ET from or to the parent molecule. We will discuss selected radical ion structure types derived from the different families of donors–acceptors to illustrate their rich variety; we will emphasize the molecular features that determine these structures.
4.1. Radical Ions of p Donors
Among the radical ions of p donors, those of aromatic hydrocarbons were the first class to be investigated in detail, because they are reasonably stable and their spectra fall into a readily accessible range, allowing them to be characterized by ESR83,84 and optical spectroscopy.58 Their structures are closely related to their parents; this manifests itself in an interesting simple relationship between the PE spectra of ground-state parent molecules and the electronic spectra of their radical cations: The excitation energies, E, of radical cations correspond approxi-
mately to differences in the ionization energies, I, of the parent molecules (cf. Fig. 6.5).85,86
Im ¼ Em |
ð15Þ |
where m are the indexes of MOs counting down from the HOMO and the index of the PE band, starting at the lowest ionization energy. The success of this simple relation is due to the fortuitous cancellation of errors inherent in the model.86
Of course, a close structural relationship between radical cations and parent molecules is not likely to hold generally, but it is a fair approximation for alternant hydrocarbons. Deviations have been noted: some stilbene radical cations have higher-lying excited states without precedent in the PE spectrum of the parent;87,88
for radical cations of cross-conjugated systems (e.g., 1) already the first excited state is without such precedent.87,89 These states have been called ‘‘non-Koop-
manns’’ states. Alkenes also feature major differences between parent and radical cation electronic structures.
• +
1• +
Of the radical ions derived from aromatic hydrocarbons, we mention the ions of benzene and tetracene. For benzene, both the positive and negative ions have been
characterized by ESR spectroscopy. The radical anion shows seven evenly spaced lines (aH ¼ 0:341 mT),90,91 suggesting that the spin density is distributed evenly
over the six carbons (or that there is a fast equilibrium between structures corresponding to the two degenerate antibonding SOMOs; Fig. 6.7). Introducing a single D is sufficient to disturb the equilibrium (aH ¼ 0:3983 mT, 4H; aH ¼ 0:3454 mT,

216 ORGANIC RADICAL IONS
Figure 6.7. Bonding and antibonding MOs of benzene.
1H; aD ¼ 0:056 mT); D shows some preference for a position in a nodal plane.92 This trend is more pronounced for a methyl group, which shows a significant
preference for the nodal plane (aH ¼ 0:5553 mT, H-2,6; aH ¼ 0:5687 mT, H-3,5; aH ¼ 0:0129 mT, H-4; aH ¼ 0:02 mT, 3H).93
The benzene radical cation (g ¼ 2:00242) also shows seven evenly spaced lines (aH ¼ 0:444 mT);94 indicating that the spin density is distributed evenly over the six carbons or that a fast equilibrium between structures corresponding to two degenerate bonding SOMOs contribute equally; Fig. 6.7). The coupling constants for the cation are slightly larger than for the anion; apparently, the nuclei interact more efficiently with an unpaired electron in a bonding than in an antibonding SOMO. A methyl group affects the hyperfine coupling pattern differently (aH ¼ 1:8 mT, Me; aH ¼ 0:21 mT, H-2,6; aH ¼ 1:18 mT, H-4): for the cation the methyl group prefers a position of high spin density rather than in the nodal plane.95
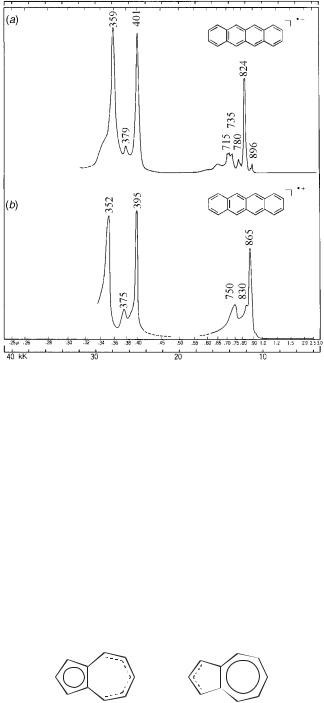
The radical ions of tetracene have been studied by ESR and optical spectroscopy. The ESR data reveal the spin density distribution; again, the splittings in the anion (aH ¼ 0:143 mT, H-1,4,7,10; aH ¼ 0:116 mT, H-2,3,8,9; aH ¼ 0:416 mT, H-5,6,11,12)96 are overall smaller than those of the cation (aH ¼ 0:168 mT, H-1,4,7,10; aH ¼ 0:102 mT, H-2,3,8,9; aD ¼ 0:501 mT, H-5,6,11,12),97 indicating that the unpaired spin in the antibonding SOMO interacts less efficiently than in the bonding SOMO. The spin densities are higher in the center and fall off somewhat toward the outer rings. The optical spectra of the anion and the cation are remarkably similar (Fig. 6.8). Such similarity is to be expected, because HOMO and
RADICAL ION STRUCTURES |
217 |
Figure 6.8. Electronic absorption spectra of the tetracene radical anion (a) and radical cation (b). [Adapted from Ref. 58.]
LUMO are ‘‘paired orbitals’’ according to a (pairing) theorem derived by McConnell and McLachlan (cf. Fig. 6.5).98 Adding an electron to or removing an electron from an aromatic hydrocarbon occurs with minimal changes in structure.
As mentioned for the relationship between the PE spectrum of a parent molecule and the electronic spectrum of its radical cation, any close correspondence between the electronic spectra of anions and cations or their hyperfine coupling patterns holds only for alternant hydrocarbons. The anions and cations of nonalternant hydrocarbons (e.g., azulene) have significantly different hyperfine patterns. Azulene
radical anion has major hyperfine splitting constants (hfcs) on carbons 6, and 4,8 (aH ¼ 0:91 mT, H-6; aH ¼ 0:65 mT, H-4,8; aH ¼ 0:38 mT, H-2);99 in contrast,
the radical cation has major hfcs on carbons 1 and 3 (aH ¼ 1: |
065 mT, H-1,3; |
||||||
|
100 |
||||||
aH ¼ 0:152 mT, H-2; aH ¼ 0:415 mT, H-5,7; aH ¼ 0:112 mT, H-6). |
|
||||||
3 |
4 |
5 |
3 |
4 |
5 |
|
|
|
|
|
|
||||
2 – |
|
• 6 |
2 • |
+ |
6 |
|
|
1 |
8 |
7 |
1 |
8 |
7 |
|
|
|
|
|
|
|
|
||

218 ORGANIC RADICAL IONS
The divergent hyperfine patterns of the two ions can be ascribed to their 6p aromatic substructures, cyclopentadienide anion, and cycloheptatrienylium cation, respectively.
4.2. Radical Ions of n Donors
Radical cations of n donors are derived typically from substrates containing one or more N, O, or S atoms; they are substituted frequently with alkene or arene moieties. Among these systems, we mention only a few examples, including two radical ions derived from 1,4-diazabicyclo[2.2.2]octane (2) and the tricyclic tetraaza
compound (3). For both ions, ESR as well as OS/PES data were measured. The bicyclic system (aN ¼ 1:696 mT, 2N; aH ¼ 0:734 mT, 12H)101 shows
perfect correspondence between the Es of the radical ion and the I’s of its precursor.102
|
• + |
• + |
• + |
|
N |
N |
|
N |
N |
||
|
|
|
|
N |
N |
N |
N |
|
|
||
|
|
|
|
2 • + |
|
3 • + |
4 • + |
In contrast, the radical cation of the tetracyclic system is significantly distorted: The parent system has D2d symmetry and a b2 HOMO, whereas the radical cation is distorted toward 2 equiv structures of C2v symmetry (2E), with a two-center three-
electron N N bond (3þ).103,104 |
The ESR data (aN |
¼ |
0:709 mT, 4N; aH |
¼ |
0:768 mT, |
|
|
|
101 |
|
|
||
8H, N C N; aH ¼ 0:414 mT, 8H, N C C) |
|
support the rapid interconversion |
||||
of the two structures. The structure of 3þ is one of many doubly or multiply bridged diaza compounds forming three-electron N N bonds (e.g., 4þ).105 Many additional examples involving three-electron S S or I I bonds are also known.106 Dioxetane radical cations (e.g., 5þ), characterized by ESR spectroscopy as intermediates in oxygenations (cf., Section 5),107 contain analogous three-electron O O bonds.
 • + 0
• + 0 0
0
5 • +
Intermolecular s-dimer cations are known for thioethers but dimeric amine cation species are not well characterized in solution. However, an example was detected in a zeolite;108 the confinement in the narrow channels and the restricted diffusion favor interaction between the two entities.
RADICAL ION STRUCTURES |
219 |
4.3. Radical Ions of r Donors
Alkanes are among the least reactive classes of compounds; they are poor electron
acceptors (low electron affinities) as well as donors (high ionization potentials, viz., CH4,12.61 eV; C10H22, 9.65 eV).109 The molecular anions of n-alkanes are espe-
cially unstable;110 negative ion yields for simple alkanes are 104 times lower
than positive ion yields. Electron attachment results in small fragment ions (CH , CH2 , CH3 , C2H ).111–113 n-Alkanes can be ionized by electron (MS) or
Hea impact (PES) in the gas phase, but oxidation in solution is difficult. Alkane radical cations have become readily accessible only with the advent of matrix isolation techniques combined with ESR detection.
Of the small alkane radical cations CH4þ has potential significance as an interstellar species and may have played a role in the chemical evolution preceding the origins of life.114 In the laboratory, however, CH4þ has long been elusive; it was finally generated by discharge ionization in a neon matrix.115 An ESR quintet
(aH ¼ 5:48 mT) suggested four equivalent protons, that is, a species with D2d symmetry. However, CH2D2þ showed a 1 : 2 : 1 triplet of 1 : 2 : 3 : 2 : 1 quintets
(aH ¼ 12:17 mT; aD ¼ 0:222 mT, corresponding to an aH ¼ 1:4 mT); nonequivalent pairs support a species with C2v symmetry. Therefore, C2v symmetry was assigned to CH4þ and CH2D2þ; the averaged geometry of CH4þ was ascribed
to dynamic Jahn–Teller (JT) distortion (Fig. 6.9).115 The C2v symmetry of CH4þ is the same as that of BH4 ,116,117 with which it is isoelectronic.
Ab initio calculations of CH4þ at the UHF/6-31G* level showed two elongated (119.6 pm; aH ¼ 13:7 mT) and two shortened C H bonds (109.4 pm; aH ¼ 1:7 mT);118 2D nuclei preferentially occupy the shorter bonds (Fig. 6.9).115 The calculated structure depends critically on the basis set and the method used to account for electron correlation. For example, the D2d structure is the global minimum using UB3LYP/6-31G*.
The ESR spectrum of C2H6þ at 4 K in SF6 shows a 1 : 2 : 1 triplet; two strongly coupled protons (aH ¼ 15:25 mT) support spin densities, r ¼ 0:3, in two H1s orbitals (s delocalization); the remaining protons are weakly coupled ( 1 mT).119 The large positive hyperfine coupling constant is reproduced well by B3LYP calculations.120 The structure of C2H6þ resembles diborane, B2H6, rather than B2H6 , with which it is isoelectronic (Fig. 6.9).121 At 77 K, the spectrum changed to a septet (a ¼ 5:04 mT, 13 of a at 4 K), indicating rapid equilibration of three
equivalent distorted forms (dynamic JT effect), with a very small activation energy, Ea ¼ 250 cal/mol.119
Propane radical cation (C3H8) gave rise to three different structures in different matrices.122 These results are ascribed to the fact that the energies of several high-lying orbitals lie close to each other, so that small perturbations due to the
|
relative energies. In SF |
, a 1 : 2 : 1 triplet (A |
¼ |
9 8 mT) |
||
matrix may alter their |
|
1 |
6 |
|
: |
|
is observed; the in-plane |
|
H nuclei at C1 and C3 are strongly coupled (s delocali- |
||||
zation).123 Compared to the parent the rotational axes of the Me groups are bent toward the central carbon and the two in-plane C H bonds are lengthened (4b1 symmetry).

220 ORGANIC RADICAL IONS
Figure 6.9. Top to bottom: Two possible Jahn–Teller distorted geometries for the methane radical cation and calculated geometry for its energy minimum;115 schematic representation of the SOMO for ethane radical cation;119 SOMOs of three different propane radical cations observed by ESR;122 the SOMO of butane radical cation;122 and SOMOs for two conformers of pentane radical cation.122
In Freon 113 two strong (10.5 mT) and four weaker hyperfine interactions (5.25 mT) were observed, assigned to a delocalized C3H8þ species with an antibonding pseudo-p orbital. The 1H nuclei at C2 are strongly coupled, the protons in the C C C plane have negligible hfcs (Fig. 6.9).122 A third structure type, obtained
in C3F8, has one strongly coupled (A ¼ 8:4 mT) and one moderately coupled proton (A ¼ 1:8 mT); it was identified as a species with one lengthened C C bond.124,125
In general, alkane radical cations are good examples of s-delocalized species. Their SOMOs are s orbitals spread over extended planar C C systems and
two in-plane 1H nuclei at the terminal carbons (Fig. 6.9). The ESR spectra show 1 : 2 : 1 triplets, due to strongly coupled terminal protons.122,126–128 The splitting
decreases from 10.5 mT for C3H8þ to 1.0 mT for C10H22þ, because the unpaired

RADICAL ION STRUCTURES |
221 |
electron spin is distributed over more and more carbon atoms. The hyperfine cou-
pling constants of the inner protons and the out-of-plane protons of the terminal methyl groups are small.123,129
The actual ESR spectra are more complex because gauche conformers are also present.122,130–134 In these species, the spin delocalization ceases at the gauche
carbon, the unpaired electron is confined mostly to the longer fragment, and the in-plane 1H nuclei are strongly coupled.122,131 For example, n-pentane exists as
an s-trans,trans,trans- and an s-trans,trans,gauche-conformer (Fig. 6.9). The extended radical cations of n-hexane (A ¼ 3:9 mT) and n-octane (A ¼ 2:9 mT) were observed exclusively inside pentasil zeolite (ZSM-5)135 enforced by the geometry of the zeolite channels.119
4.4. Radical Ions of Strained Ring Compounds
Pronounced differences between radical cation structures and their parents must be expected for strained ring compounds. The HOMO or LUMO of these systems may be localized mainly in one bond, which may be weakened or actually break upon ionization. The oxidation potentials of strained ring compounds are lower than those of unstrained substrates because strain energy is released, resulting in noticeable changes in structure.
The potential energy surfaces of radical cations may differ from those of their neutral diamagnetic parents in three features: (1) reduced reaction barriers;
(2) reduced or reversed free energy differences between isomers; and (3) energy minima on the radical cation surface may have geometries corresponding to transition structures on the parent potential surface. We will discuss a range of structures, focusing on the unusual ones.
Cyclopropane (D3h symmetry) has a degenerate pair of in-plane e0 orbitals (S, A). Vertical ionization leads to a doubly degenerate 2E0 state, and JT distortion results in two nondegenerate electronic states, 2A1 and 2B2 (C2v symmetry),136 corresponding to two different molecular structures. The 2A1 state (orbital S singly occupied) corresponds to a structure with one lengthened C C bond; it is lowest in energy for many cyclopropane radical cations (Fig. 6.10).
An ESR spectrum at 4.2 K shows two strongly coupled 1H nuclei (aH ¼ 2:04 mT,
b protons) and four less strongly coupled ones (aH ¼ 1:17 mT, a protons);137 at 77 K, a single line was observed.138,139 The low temperature spectrum supports the
2A1 structure; at higher temperature, three equivalent 2A1 structures are averaged by dynamic JT distortion. The 2B2 structures are transition states; ab (2.04 mT) and aa ( 1.17 mT) fortuitously average to aavg 0. A ring-opened structure, 7þ, was observed in CF2Cl-CFCl2 matrices.140
|
|
• + |
• + |
H2C • |
+ CH2 |
|
||
6 • + |
7 • + |
8• + |

222 ORGANIC RADICAL IONS
Figure 6.10. The degenerate pair of cyclopropane HOMOs, S and A (top) and schematic radical cation structures, type A and type B, of states 2A1 and 2B2, respectively, generated by removing an electron from one of these orbitals (bottom).
Ab initio calculations (UHF/6-31G*//MP2/6-31G*) on the C3H6þ potential surface confirm the 2A1 structure, but fail to support the existence of 7þ.141,142 The
conversion of 6þ (2A1) to propene radical cation, 8þ ( 10 kcal/mol below 6þ), has a barrier of 30 kcal/mol (UMP2/6-31G*),143 approximately one-half that measured for cyclopropane itself.144 In contrast, 7þ rearranges to 8þ without a chemically significant barrier.142 In the light of these results the observation of 7þ at cryogenic temperatures140 must be ascribed to matrix forces (cf. the propane
radical cations).
Because radical ions of structure type A are well established,140,145,146 examples of an alternative structure are of special interest. Substitution at two carbons should stabilize the S orbital (2A1), whereas substitution at a single carbon should stabilize the A HOMO (2B2) by conjugation, homoconjugation, or hyperconjugation.147
According to ab initio calculations on methyland 1,1-dimethylcyclopropane,148 hyperconjugation falls short of stabilizing type B structures; they are transition states, undergoing second-order JT-type distortions to unsymmetrical structures with one very long C C bond. The hyperfine coupling constants (a2 ¼
1:43 mT; a3 ¼ 1:98 mT; B3LYP/6-31G*//MP2/6-31G* calculations) support a structure of type A. However, conjugation with a vinyl149 or phenyl group,150,151
or homoconjugation in norcaradiene and derivatives152 are sufficient to stabilize type B structures.
Phenylcyclopropane radical cation (9þ) has divergent hyperfine coupling constants for the secondary cyclopropane protons (abtrans ¼ 0:78 mT; abcis ¼0:07 mT; CIDNP, B3LYP/6-31G* calculations), apparently because the cis protons are located in a nodal plane.150 Similarly, vinylcyclopropane radical cation is

RADICAL ION STRUCTURES |
223 |
stabilized by conjugation; it has two conformers, s-anti-10þ and s-syn-10þ; both are type B structures, resembling a p complex between vinylmethylene and ethene.149 Interestingly, the array, Cb Ca C1, has been converted, in essence, to an allyl moiety.
Hα |
• + |
• + |
• + |
|
|
Htr |
Htr |
|
|
|
|
|
Hβ,tr |
Hcis |
Hcis |
|
|
||
|
|
|
|
|
Hβ,cis |
|
|
9 • + |
|
anti-10 • + |
syn-10 • + |
For radical cations of norcaradiene and derivatives,152 the interaction of the cyclopropane in-plane e0 orbitals with the butadiene frontier MO favors the type B structure. The assignments are based on ab-initio calculations, CIDNP results, and the ET photochemistry. The norcaradiene radical cation (11aþ) has a 2A00 electronic ground state (Cs symmetry). The C1 C6 bond is shortened on ionization ( 3.4 pm) while the lateral bonds are lengthened (þ2.8 pm). The delocalization
of spin density |
to C7 (r7 ¼ 0:246; r2;5 ¼ 0:359) |
and the hyperfine coupling |
|||||
constants of the |
cyclopropane moiety (a1;6 |
¼ |
1:36 mT; a7syn |
¼ |
0:057 mT; |
||
|
|
|
|||||
a7anti ¼ 0:063 mT) support a type B structure. |
152 |
|
|
|
|||
|
|
|
|
|
|||
The assignment of an antisymmetrical cyclopropane SOMO to the radical cation (12þ) of benzobicyclo[4.1.0]hepta-2,4-diene rests on CIDNP effects, particularly on characteristic differences to those for cis-1,2-diphenylcyclopropane (Fig. 6.11).145 The calculated carbon spin densities of 12þ on C2 (r2 ¼ 0:355), C5 (r5 ¼ 0:153), and C7 (r7 ¼ 0:149) and negative spin densities on the tertiary cyclopropane carbons (r1 ¼ 0:009, r6 ¼ 0:007)152 are in qualitative agreement with the CIDNP effects.146
For tricyclo[4.3.1.01,6]deca-2,4-diene radical cation (11bþ, the spin density
on the bridge carbon |
(r10 ¼ 0:203, |
r2;5 ¼ 0:383; a10syn ¼ 0:54 mT; a10anti ¼ |
|||||
0:48 mT) support a type B structure (UHF/6- |
31G*; C |
s |
symmetry imposed) and |
||||
|
152 |
|
|||||
a noticeable contribution due to homoconjugation. |
|
|
|
||||
7 |
• + |
10 |
• + |
|
|
|
• + |
5 |
5 |
|
|
|
|
|
|
2 |
2 |
|
|
|
|
|
|
11a • + |
11b • + |
|
|
|
|
12 • + |
|
The case of cyclobutane radical cation presents another interesting structural problem. The parent has a puckered ring with D2d symmetry; ET from one of its
e orbitals leads to a JT unstable radical cation, which distorts to structures of D2d and C2v symmetry.153 The ESR spectrum at 4 K (aH ¼ 4:9 mT, 2H; aH ¼ 1:4 mT,