

Reactive Intermediate Chemistry
.pdfBASICITY OF CARBANIONS–ACIDITY OF CARBON ACIDS |
81 |
well known that ring strain causes an increase in the amount of s character in the C H bonds attached to the ring in order to accommodate the greater p character needed for the small angle C C bonds. This enhances their acidity because the carbanion’s lone pair uses an orbital that is effectively more electronegative—more s character leads to a more compact orbital that places electron density nearer to the nucleus. To illustrate this point, Natural Bond Order (NBO) analysis from MP2 calculations indicates that the lone pair of the carbanion from cyclopropane has 34% s character, as opposed to 25% in a perfect sp3 hybrid.106 This effect does not play a significant role in cyclobutane (only 24% s character in the carbanion lone pair) and the four-membered ring compound exhibits an acidity that is consistent with a system containing secondary carbons. A similar argument can be developed using the NMR 13C–1H coupling constants of the ring C H bonds to estimate the %s character of the bonding hybrid (32% for cyclopropane and 27% for cyclobutane).109 Moreover, Streitwieser et al.110 showed that the 13C–1H coupling constants correlate with the kinetic acidities of a series of cycloalkanes.
When assessing the effect of structure on the stability of more complex carbanions derived from carbon centers that are initially sp3 hybridized, there are two general classes of species to consider. In the first case (4), the carbanion can be viewed (at least to a rough approximation) as a localized charge center that is stabilized by a substituent that acts a dipole, a polarizable group, an electronwithdrawing group, or some combination of these roles. These systems can be analyzed within the fundamental framework of substituent effects. In the second case (5), the carbanion is part of a cyclic p system and the charge is dispersed equally or nearly equally over several carbons. Here, the nature of the p system plays a critical role in the stability of the carbanion though substituents attached to the p-system can also play important roles.
|
|
H |
|
|
C |
Z CH |
– |
– |
|
||
2 |
|
|
45
We will begin with the first group of species and then move to the second. To initially avoid a discussion of solvation effects on carbanion stability, we will begin with gas-phase data for simple representatives of model classes of compounds (Table 3.2). Later, the gas-phase data will be compared to data that have been obtained in solution (Section 4.3.3).
When a substituent, Z, is added to a carbanion, it can have three important modes of stabilizing the anion. First, if it is more electronegative than carbon, it can inductively stabilize the carbanion by shifting electron density along the C Z s bond thereby effectively reducing the charge on the carbon (6). Second, if the substituent has low-lying p orbitals it can conjugate with the carbon lone pair and disperse the charge through a p-bonded resonance form (7). Third, the substituent can polarize its own electron density thereby creating an effective dipole
82 CARBANIONS
TABLE 3.2. Gas-Phase DHacid Values for
Me Z Compoundsa
ZHacid
H |
417 |
F |
409 |
Cl |
400 |
Br |
397 |
OMe |
407 |
SMe |
390 |
PMe2 |
385 |
SiMe3 |
388 |
S(O)Me |
374 |
S(O)2Me |
366 |
CH CH2 |
389 |
Ph |
381 |
C CH |
381 |
C(O)Me |
369 |
C N |
373 |
NO2 |
357 |
a In kilocalories per mole (kcal/mol.) (see Ref. 54).
(or enhancing one that is already present in the neutral carbon acid) to stabilize the carbanion center (8).
δ– |
δ– |
|
|
|
– |
– δ+ |
δ– |
|
H2C |
Z |
H2C |
|
|
Z |
H2C |
Z |
|
|
|
|||||||
7 |
||||||||
|
6 |
|
|
8 |
|
|||
In Table 3.2, gas-phase Hacid values are given for a group of substituted methane derivatives. The first few entries involve the addition of a halogen to the carbon acid. As expected, these electron-withdrawing groups inductively stabilize the resulting carbanion and therefore increase the acidity. Counter to the variation in electronegativities, the stabilizing effect on the carbanion increases along the series F < Cl < Br. This trend highlights the fact that the larger halogens are more polarizable. A similar, but more dramatic trend is seen in the data for oxygen and sulfur substituents (OMe and SMe) where the latter provides 17 kcal/mol more stabilization than the former. Obviously, polarizability is playing a much greater role than electronegativity in these gas-phase stabilizations. Another factor that Wiberg and Castejon111 have highlighted in this system is negative hyperconjugation.37,112 Here, conjugation of the carbon lone pair with a s* orbital on sulfur leads to charge dispersal and stabilization (10).
|
S |
– |
|
CH – |
S |
H C |
CH |
|
CH |
||
|
|||||
3 |
2 |
3 |
2 |
||
|
9 |
|
|
|
10 |
BASICITY OF CARBANIONS–ACIDITY OF CARBON ACIDS |
83 |
Using computational methods, Wiberg showed that this effect is only significant with sulfur, not oxygen substitution. This mode of stabilization is in effect, a resonance description of a type of polarization in that electron density is shifting in the substituent group in response to the carbanion. However, negative hyperconjugation also indicates the formation of a partial p bond to the sulfur. The entries for the other third period elements (phosphorous and silicon) also highlight the importance of polarizability in the gas phase. Although there is a significant variation in electronegativity across the series Si < P < S < Cl, there is a relatively small variation in the stabilizing effects with the most electronegative element, chlorine, providing the least stabilization to the carbanion. Here, it appears that the additional Me groups used to fill-out the valencies of the third period elements in moving from right to left across the periodic table (i.e., MeS vs. Me2P vs. Me3Si) may provide additional polarizability that counters the effects of electronegativity. Of course, negative hyperconjugation could also be playing a role with phosphorous and silicon. The next two entries in the table involve sulfoxide and sulfone substituent groups. Both are highly polar with large internal dipole moments and therefore act as powerful stabilizing groups via inductive and through-space field effects (i.e., the interaction of the S O dipole with the carbanion center). These groups lead to much more stable carbanions than those stabilized by the halogens or a simple sulfur group (SMe).
The rest of the entries in the table have p systems to conjugate with the carbon lone pair and therefore offer additional resonance forms for the carbanion. The vinyl and phenyl substituents are weakly polar and their effect is mainly from resonance delocalization along with polarizability. The greater stabilizing effect of the phenyl group is probably a result of its larger size and therefore greater polarizability. The ethynyl substituent is about as stabilizing as a phenyl group and this is probably related to the unusually high electronegativity of an sp-hybridized carbon (see Section 4.2.2 for more discussion of this effect). The final entries in the table are for species that allow for resonance forms where the carbanion’s charge is carried by a more electronegative atom, oxygen or nitrogen (evidence for resonance delocalization onto nitrogen is not conclusive, see below).
|
|
O– |
|
|
|
|
|
O |
– |
|
|
|
|
|
H C |
|
C |
|
N – |
H C N + |
|
H C |
|
CCH |
|
|||||||
|
|
|
|
|||||||
|
|
|
|
|||||||
2 |
3 |
2 |
|
|
|
|
2 |
|
||
O–
As a result, these anions can gain exceptional stability and their carbon acids are unusually acidic. Because the above resonance forms probably play very important roles in stabilizing the anion, these species are not purely carbanions. However, thermodynamics favors reactions (protonation or alkylation) at carbon so their behavior is generally characteristic of carbanions. It also should be noted that
kinetics often favor reaction at the heteroatom in these anions though leading to the less stable product.113–115
In Table 3.3, data are given for some polysubstituted methane derivatives. Here it is possible to judge the effects of sequential additions of the same substituent. The

84 CARBANIONS
TABLE 3.3. Gas-Phase DHacid Values for
Polysubstituted Methanesa
Species |
Hacid |
(Subst)b |
H3CCl |
400 |
17 |
H2CCl2 |
376 |
24 |
HCCl3 |
358 |
18 |
H3CPh |
381 |
36 |
H2CPh2 |
364 |
17 |
HCPh3 |
359 |
5 |
CH3C(O)Me |
369 |
48 |
H2C[C(O)Me]2 |
344 |
25 |
HC[C(O)Me]3 |
329 |
15 |
CH3C N |
373 |
44 |
H2C(C N)2 |
336 |
37 |
a In kilocalories per mole (kcal/mol) (see Ref. 54).
b Effect of additional substituent. In monosubstituted system, difference from methane.
first entries involve the addition of chlorines. It can be seen that to a rough approximation, each chlorine has a similar acidifying effect on the carbon acid. In contrast, the acidifying effect of additional phenyl groups drops off dramatically and the third one in the triphenylmethyl anion has only a marginal effect. The difference between these two systems can be linked to how they stabilize the carbanion. It appears that inductive stabilization (i.e., the Cl system) is not affected by the presence of additional substituents (however, this pattern was not observed in the inductive destabilization of carbanions by methyl groups, Section 4.2.1). In contrast, resonance stabilization (i.e., the phenyl system) is deeply attenuated by other conjugating substituents. A rational explanation is that each conjugating substituent reduces the charge density on the carbanion, leaving less density to be delocalized by the next conjugating substituent.116 Of course, steric effects also play a role as the poly-substituted systems become crowded making conjugation and optimum orbital overlap difficult. A similar trend is seen with the ketone substituent, which is also expected to be dominated by resonance effects. However, the drop-off is not as dramatic as with the phenyl group hinting that dipole effects play a significant role with this substituent. The final entries refer to the cyano group. Data are available for monoand dicyanomethane and the acidifying effect of the second group nearly matches the first. This suggests that although resonance stabilization is possible with the cyano group, it acts mainly through inductive and field effects, which are diminished to a lesser extent by polysubstitution. Data from aqueous solution also support this conclusion.117
Conjugated ring systems offer an alternative mode for the stabilization of a carbanion center. The most common situation is where deprotonation completes a cyclic p system leading to a highly stabilized, aromatic anion. The best known example is cyclopentadiene, which leads to a six-electron, aromatic ring after
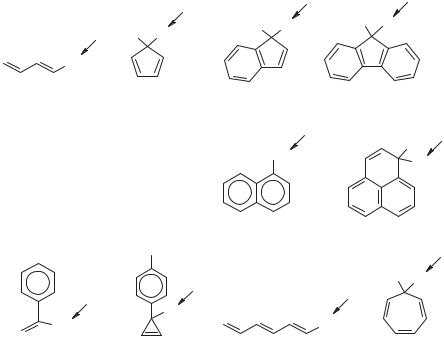
BASICITY OF CARBANIONS–ACIDITY OF CARBON ACIDS |
85 |
deprotonation. The Hacid value for cyclopentadiene, 354 kcal/mol, is much lower than expected for a carbanion conjugated to a simple polyene system (Fig. 3.1). For example, pentadiene has a Hacid value of 369 kcal/mol indicating that aromaticity provides roughly 15 kcal/mol of stabilization to the carbanion. To put this energy difference in perspective, it would translate to a difference of >10 units in terms of pKa values at 300 K. Indene and fluorene represent larger analogues and also display unusually high acidity with Hacid values near that of cyclopentadiene. Here, larger size and greater polarizability do not lead to a significant acidity enhancement suggesting that fusing the additional aromatic rings onto the cyclopentadiene framework has a mild acid weakening effect that balances the increased polarizability. This topic will be discussed in more detail in Section 4.3.3. Phenalene offers another example of a system where deprotonation completes a p system and in this case, produces a 14-electron, aromatic carbanion. Unlike the cyclopentadiene derivatives, it is not possible to draw out resonance forms for the phenalenide anion where each of the ring carbons bears the charge. Nonetheless, phenalene has aHacid value that is comparable to cyclopentadiene and much smaller than that
|
H H |
H H |
H H |
|
|
|
|
|
|
CH3 |
|
|
|
|
1,3-Pentadiene |
Cyclopentadiene |
Indene |
Fluorene |
|
∆Hacid = 369 |
∆Hacid = 354 |
∆Hacid = 352 |
∆Hacid = 352 |
|
|
|
CH3 |
H |
|
|
|
|
H |
|
|
|
|
|
|
|
|
1-Methylnaphthalene |
Phenalene |
|
|
Me |
∆Hacid = 374 |
∆Hacid = 351 |
|
|
|
|
H |
H |
|
|
|
|
|
|
H |
|
|
|
CH |
|
CH3 |
|
|
3 |
|
|
|
|
2-Phenylpropene |
3-(p-Tolyl)cyclopropene |
1,3,5-Heptatriene |
Cycloheptatriene |
|
∆Hacid = 386 |
∆Hacid = 399 |
∆Hacid = 365 |
∆Hacid = 375 |
|
Figure 3.1. Gas-phase Hacid values (kcal/mol) for carbon acids that give aromatic or antiaromatic carbanions.54 Acyclic analogues are included for comparison. Arrow indicates acidic hydrogen.

86 CARBANIONS
of 1-methylnaphthalene, an analogue that contains some, but not all of the stabilizing features of phenalene (i.e., it lacks one double bond).
Antiaromaticity also plays a role in the stability of carbanions. In Figure 3.1, examples of fourand eight-electron antiaromatic systems are provided. Kass and co-workers118 determined the gas-phase acidity of a 3-arylcyclopropene derivative and its Hacid value is much higher than that for a typical allylic anion. Compared to 2-phenylpropene, the ring system destabilizes the carbanion by 13 kcal/mol. One could also compare the cyclopropene system to a saturated analogue, phenylcyclopropane, and that suggests that antiaromaticity destabilizes the anion by 9 kcal/mol. Neither of these is a perfect analogue because the former lacks the acidifying effect of a three-membered ring structure and the latter lacks allylic conjugation to the carbanion. In any case, these analogies suggest that the cyclopropenyl anion’s stability is reduced due to antiaromaticity. The effects of antiaromaticity are also seen in the deprotonation of cycloheptatriene, which leads to an eight-electron, antiaromatic system and has a Hacid value that is considerably higher than that of an acyclic analogue, 1,3,5-heptatriene. Both of these examples highlight the fact that antiaromaticity as well as aromaticity play significant roles in the stability of carbanions.
4.2.2. Carbanions Derived from sp2 and sp Hybridized C H Bonds. In the discussion of carbanions derived from cycloalkanes, it was noted that increased s character in the carbon lone pair leads to a more stable carbanion. A similar effect is expected in carbanions derived from sp2 and sp hybridized C H bonds. In Table 3.4, gas-phase data are given for a variety of unsaturated systems. The experimental values for these species are generally very accurate and it can be seen that in most cases, the computed values match very well in these systems. One exception is ethyne, where a range of computed values has been reported, both higher and lower.119 However, high-level calculations on ethyne do give a value close to experiment.120 It can be seen that in going from ethane (Table 3.1) to ethene, theHacid value drops 11 kcal/mol indicating significant stabilization is gained in the
TABLE 3.4. Gas-Phase DHacid Values of sp and sp2 Hybridized
Carbon Acidsa
Carbon Acid |
Hacid (exp)b |
Hacid (comp)c |
||
H C CH |
409 |
408 |
||
|
2 |
2 |
402 |
d |
c-C6H6 |
|
402 |
||
HC CH |
|
378 |
372e |
|
H2C C CH2 |
381 |
383 |
||
H2C C O |
365 |
366 |
||
a In kilocalories per mole (kcal/mol). |
|
|||
b See Ref. 54. |
|
|
||
c |
Values from CCSD(T)/6-311 |
þ þ G(d,p) calculations (see Ref. 121). |
||
d |
||||
e |
Values from MP2/6-31 þ G(d,p) calculations (see Ref. 122). |
|
||
|
Values from MP2/aug-cc-pVDZ calculations (see Ref. 123). |
|
||

BASICITY OF CARBANIONS–ACIDITY OF CARBON ACIDS |
87 |
sp2 hybridized carbanion. For comparison, the effect of the hybridization change in going from propane (2 carbon) to cyclopropane leads to a 7 kcal/mol increase in acidity (Table 3.1). Given that the NBO calculations suggested that the cyclopropyl anion had nearly sp2 hybridization in its lone pair (34% s character),106 it is not surprising that a similar acidifying effect is seen in going from ethane to ethene. Benzene provides another example of an sp2 hybridized carbon acid and itsHacid value is somewhat smaller than ethene because it is a larger, more polarizable system. The shift to an sp hybridized system leads to a dramatic increase in acidity. Ethyne’s Hacid value is 31 kcal/mol below that of ethene. A large effect is expected because the system is going from roughly 33% s character in ethene to 50% in ethyne, a much larger change than in going from ethane to ethene. This trend is also seen in the C H homolytic bond energies and bond distances across the ethane, ethene, ethyne series. Allene presents a special case because the resulting carbanion has a resonance form that is best described as an ethynyl stabilized carbanion (propargyl system). This resonance form and the resulting delocalization explain the relatively high acidity of allene compared to ethene. As expected, ketene is also unusually acidic for an sp2 hybridized carbon acid and again, this results from the presence of a highly stable resonance form in the anion, an oxide in this case.
– |
|
|
|
|
|
|
|
|
|
|
|
– |
||
HC |
|
|
C |
|
|
CH2 |
|
HC |
|
C |
|
CH2 |
||
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
||||||||
– |
|
|
|
|
|
|
|
|
|
|
|
– |
||
HC |
|
|
C |
|
|
O |
|
HC |
|
C |
|
O |
||
|
|
|
|
|
|
|
||||||||
4.3. Condensed-Phase Carbon Acidity Measurements
Carbon acidities have been measured in several solvents, but the greatest amount of data has been accumulated in aqueous mixtures,124,125 DMSO,38,39 cyclohexyl- amine,40–47 and various ethers such as DME52,53 and THF.48–51 These solvents offer
different limitations and environments for the resulting carbanions. The high dielectric of water (78.5) leads to the formation of free-ions, but its relatively low pKa (15.5) limits its use to exceptionally strong carbon acids due to the leveling effect (see above). DMSO also has a high dielectric (46.7) so free ions are common, but it has a much higher pKa (35.1) than water so a wider range of species may be studied in it. Cyclohexylamine is not nearly as polar as the former solvents and leads to ionpair formation for carbanion salts; however, its low acidity (cesium ion pair pKa ¼ 41:6) allows for the study of weaker acids than DMSO. Finally, the ethers also generally lead to ion-paired carbanion salts and have high effective pKa values. The ethers do not yield stable carbanions when deprotonated, but instead, decompose. Therefore, the solvent cannot be used as an absolute standard and the effective pKa is essentially a measure of the kinetic stability of the solvent towards highly basic carbanions. In this section, no effort will be made to present a comprehensive listing of all the measurements completed in solution. Many workers have made important contributions in a variety of solvents and the data can be found in

88 CARBANIONS
the original references and reviews.23,37,38,125 Instead, general trends will be presented, free ion and ion-pair acidities will be contrasted, and comparisons to the gas phase will be made.
4.3.1. Carbon Acidity in DMSO. Although DMSO has a high dielectric constant, it is unable to stabilize anions via hydrogen bonding. Consequently, anions that are capable of strong hydrogen-bonding interactions are much more difficult to form in DMSO than water. On the other hand, anions that are diffuse or highly delocalized, a common situation with carbanions, are not as sensitive to the hydrogen bonding ability of the solvent and are nearly as stable in DMSO as in water. This relationship is illustrated in Table 3.5 with data from Bordwell’s group for measurements made in DMSO at 25 C. For compounds that give small, highly localized anions like HCl and MeOH, there is a very large difference between the water and DMSO values (10–15 units), but for more delocalized systems the difference is much smaller. For example, the pKa of malononitrile [H2C(C N)2] is the same in water and DMSO, which suggests that for carbanions in general, the DMSO values are reasonably close to what might be expected in an aqueouslike environment.
Table 3.6 contains a sample of pKa values determined in DMSO for a variety of common functional groups. These were determined in Bordwell’s group by measuring equilibria between carbon acid pairs (i.e., Eq. 5) and then linking the resulting scale to the absolute pKa of the solvent. It can be seen that highly polar substituents capable of conjugation such as the nitro, carbonyl, or cyano group lead to pKa values in the range from 17–31. In other words, carbanions stabilized by a single substituent, no matter how potent, are very basic in DMSO solution. Multiple acidifying groups are needed to bring carbanions into the pKa range where any significant self-ionization might occur. For example, malononitrile and 1,3-pentadione have pKa values in DMSO that are <16 (11.0 and 13.3, respectively). In the absence of polar substituents, resonance stabilized carbanions have pKa values that are >30. For example, the series from toluene to triphenylmethane have pKa values from 30.6– 43. In contrast, systems that lead to aromatic anions have significantly lower pKa values. Cyclopentadiene has a pKa of 18 and its larger derivatives, indene and fluorene, have pKa values of 20.1 and 22.6, respectively. Finally, sp hybridization leads to a significant increase in acidity with phenylacetylene exhibiting a pKa of
TABLE 3.5. The pKa Values Measured in Water and DMSOa
Speciesb |
pKa(water) |
pKa(DMSO) |
HCl |
8.0 |
1.8 |
MeSO3H |
0.6 |
1.6 |
MeCO2H |
4.8 |
12.3 |
MeC(O)CH2C(O)Me |
8.9 |
13.3 |
H2C(C N)2 |
11.0 |
11.0 |
MeOH |
15.5 |
29.0 |
a See Ref. 38.

BASICITY OF CARBANIONS–ACIDITY OF CARBON ACIDS |
89 |
TABLE 3.6. Measured pKa Values in DMSO for
Representative Functional Groupsa
Speciesb |
pKa |
CH3NO2 |
17.2 |
CH3C(O)CH3 |
26.5 |
CH3C(O)OEtc |
27.4 |
CH3SO2CH3 |
31.1 |
CH3C N |
31.3 |
CH3S(O)CH3 |
35.1 |
Cyclopentadiene |
18.0 |
Indened |
20.1 |
Fluorened |
22.6 |
PhC CH |
28.7 |
Ph3CH |
30.6 |
Ph2CH2 |
32.2 |
PhCH3 |
43e |
|
|
a In kilocalories per mole (kcal/mol) (see Ref. 38). b Site of deprotonation is in bold.
c See Ref. 126.
d See Figure 3.1 for structures. e Estimated value.
28.7, considerably lower than the resonance stabilized, sp3 hybridized hydrocarbon systems like toluene.
The data in DMSO also allow for an evaluation of the effect of a-alkyl groups on the stability of a carbanion. Because alkyl groups are generally categorized as electron donors, one might expect that they would destabilize a carbanion, but the data indicate that the situation is not so simple. In Table 3.7, pKa and Hacid values are
TABLE 3.7. Effect of a-Methyl Groups on the Acidity of Carbon Acids in DMSO and the Gas Phase
Speciesa |
pKa(DMSO)b |
Hacidc |
PhSO2CH3 |
29.5 |
362.7 |
PhSO2CH2Me |
31.3 |
365.0 |
CH2(C N)2 |
11.4 |
|
MeCH(C N)2 |
12.4 |
|
CH3NO2 |
17.7 |
356.4 |
MeCH2NO2 |
17.0 |
355.9 |
Me2CHNO2 |
16.9 |
356.0 |
Fluorene |
22.9 |
351.7 |
9-Methylfluorened |
22.3 |
350.8 |
a Site of deprotonation is in bold.
b See Ref. 127. Reported on a per acidic hydrogen basis in this table. c In kilocalories per mole (kcal/mol) (see Ref. 54).
d The 9-position is the point of deprotonation.
90 CARBANIONS
given for three sets of homologues that differ only by the number of methyl groups attached to the carbanion center. In the first two sets, phenylsulfone and malononitrile derivatives, the addition of an a-methyl group causes a significant decrease in the acidity (destabilizes the carbanion). This result is most likely from the aforementioned electron-donating capabilities of the methyl groups. Because each of these systems is expected to give a fairly localized carbanion, the effect is maximized. The destabilizing effect of a methyl group was also seen in the gas phase in going from methane to ethane (Section 4.2.1). For nitro-stabilized carbanions, one sees the opposite effect, a weak, but general increase in acidity as methyl groups are added to the a carbon. This effect is also observed with fluorene (see Fig. 3.1 for the structure) and its 9-methyl derivative. In both of these systems, deprotonation leads to an sp2 hybridized carbanion with extensive charge delocalization into other parts of the molecule. It is well known that alkyl substituents stabilize sp2 hybridized centers (i.e., more highly substituted alkenes are more stable). Bordwell et al.127 argued that this effect counters the inductive effect of the methyl group and renders it mildly stabilizing for a carbanion. From the data in Table 3.7 as well as the data presented earlier on the gas-phase acidity of alkanes (Section 4.2.1 and Table 3.1), it is clear that alkyl groups do not have a uniform effect on the stability of a carbanion center and systems have to be analyzed on an individual basis taking into account polarizability as well as inductive, hybridization, and steric effects.
Finally, Arnett et al.128–130 developed a different approach to determining the stability of carbanions in DMSO. By means of solution calorimetry, the heat of protonation of a carbanion can be assessed in this solvent. As one might expect, there is a good correlation between the relative heats of protonation measured in this way and relative free energies of protonation measured by Bordwell using the methods described above.130
4.3.2. Ion-Pairing and Carbon Acidity. Many studies of carbon acidity have been completed in solvents such as ethers that favor the formation of ion-paired carbanions. In these cases, the equilibrium measurements (Eq. 6) reveal the relative stabilities of the metal salts of the carbanions. If the interaction of the metal cation with the carbanion is weak or at least uniformly strong across a group of compounds, then the ion-pair acidities will parallel those observed in true pKa measurements. On the other hand, if there are strong, specific interactions in the salts, then the carbanion may be preferentially stabilized causing its parent compound to appear unusually acidic. In Figure 3.2, the ion-pair acidities in THF and cyclohexylamine (CHA) of a series of hydrocarbons (e.g., cyclopentadiene and fluorene derivatives as well as diand triphenylmethane) are plotted against the pKa values in DMSO. Independent of the solvent (THF or CHA) or counterion (Liþ or Csþ), there are good correlations between the ion-pair and free-ion values. Clearly, the ionpairing energies in the hydrocarbon derived carbanions are relatively constant and therefore do not affect the acidity measurements. The point that falls far below
the correlation line is not for a hydrocarbon, but instead is for a ketone, p-biphenyl isopropyl ketone. Streitwieser and co-workers131,132 determined lithium and cesium