

Cell Biology Protocols
.pdfPROTOCOL 6.13 |
251 |
with limited solubility in water and with low lipophilicity in a lipid bilayer coat.
Technical principle
Repeated freezing and thawing of a concentrated aqueous solution of cisplatin in the presence of anionic phospholipids result in the formation of cisplatin nanocapsules. The preparation of cisplatin nanocapsules requires the presence of negatively charged phospholipids and of positively charged aqua-species of cisplatin, pointing to an essential role for electrostatic interactions in the mechanism of formation. A solution of cisplatin in water, in the absence of added chloride, contains a mixture of the neutral dichlorideand dihydroxo-species of cisplatin with low solubility in water, and positively charged aqua-species of cisplatin with a much higher solubility [5]. In the model proposed for the mechanism of nanocapsule formation [9], cisplatin is concentrated in the residual fluid during freezing, forming small aggregates when the solubility limit of the dichloro-species is exceeded. As freezing proceeds, the nanoprecipitates of the dichloro-species of cisplatin become covered by the positively charged aquaspecies, which have a higher solubility limit. The negatively charged membranes interact with the positively charged cisplatin aggregates and then reorganize to wrap the aggregates in a phospholipid bilayer coat. The resulting nanocapsules do not redissolve upon thawing.
Procedure
1. Cisplatin is dissolved in MilliQ water
to |
a concentration of 5 mM, |
which |
is |
facilitated by incubating at |
55 ◦C |
for 30 min. The solution is incubated overnight in the dark at 37 ◦C to ensure full equilibration.
2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) are prepared in chloroform (concentration 5 mM). The precise concentrations are determined by phosphate analysis [10].
3.Aliquots corresponding to 0.6 micromole of each phospholipid are mixed, the solvent is removed by rotatory evaporation, and the lipid film is further dried under vacuum overnight.
4.The dry lipid film is hydrated by adding 1.2 ml of the 5 mM cisplatin solution
in water and incubating for 15 min at 37 ◦C.
5.After brief homogenization on a vortex
mixer, the dispersion is transferred to a glass tube and subjected to 10 freeze–thaw cycles using ethanol/dryice (−70 ◦C) and a water bath (37 ◦C).
6.The resulting colloidal solution is transferred to microfuge tubes and centrifuged for 4 min at 470g (2100 rpm in an Eppendorf centrifuge) to collect the nanocapsules.
7.After removal of the supernatant, the fluffy white layer on top of the yellow pellet, corresponding to large liposomes, is removed by a micropipette. The yellow pellet containing the cisplatin nanocapsules, is resuspended in 1 ml water and centrifuged as above in order to wash away non-encapsulated cisplatin.
8.Upon resuspending the final pellet in
0.5 ml water, the nanocapsules are stored at 4 ◦C until use.
Alternatively, the nanocapsules are separated from contaminating liposomes by density gradient centrifugation. Briefly, the dispersion obtained after the freeze–thaw cycles is loaded on top of a step gradient consisting of 1 ml of each 1.8, 0.6
2.Stock solutions of 1,2-dioleoyl-sn-gly- and 0.2 M sucrose in 10 mM Pipes-NaOH, cero-3-phosphocholine (DOPC) and 1, 1 mM EGTA, pH 7.4. After centrifugation
252 IN VITRO TECHNIQUES
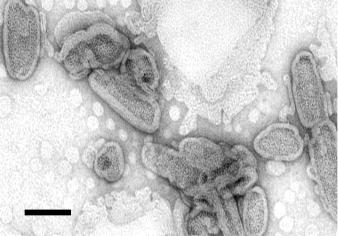
Figure 6.9 Electron micrograph of cisplatin nanocapsules visualised by negative staining. A dilute suspension of nanocapsules was transferred to a carbon-formvar coated grid, and stained with 4% (w/v) uranyl acetate for 45s. Scale bar, 100 nm
at 4 ◦C for 30 min at 400 000g, the pellet fraction corresponding to the nanocapsules is collected and washed as above.
Instead of DOPS, other anionic phospholipids like dioleoyl-phoshatidylglycerol (DOPG) and dioleoyl-phosphatidic acid (DOPA) can be used to prepare cisplatin nanocapsules. DOPC can be replaced by dioleoyl-phosphatidylethanolamine (DOPE) or sphingomyelin. The method is sensitive to high chloride concentrations and alkaline pH, because these conditions prevent the formation of the positively charged aqua-species. Instead of hydrating a lipid film with 5 mM cisplatin, it is also possible to add the cisplatin solution to preformed DOPC/DOPS liposomes and then start the freeze–thaw cycles. The cisplatin nanocapsules can be stored after lyophilization, and retain cisplatin upon rehydration.
Characterization
1. Encapsulation efficiency
The phospholipid content of the nanocapsules is determined by phosphate analysis [10]. The cisplatin content of the
nanocapsules is assessed by flameless atomic absorption spectrometry (NFAAS) using K2PtCl2 as a standard [11]. Analysis of the cisplatin nanocapsules prepared according to the above protocol, typically yields a Pt/phosphate molar ratio of 11 ±
1.Based on the size of the nanocapsules (see below), this number is estimated to correspond to an internal cisplatin concentration exceeding 0.5 M, which is far beyond the solubility limit of cisplatin and consistent with the quasi-crystalline structure of the encapsulated cisplatin [9]. The method allows encapsulation of cisplatin with an efficiency of approximately 30%.
2.Shape and size
Analysis by negative stain electron microscopy reveals bean-shaped particles consisting of an electron-dense core surrounded by a bright layer (excluding stain), corresponding to the bilayer coat (Figure 6.9). The nanocapsules have a heterogeneous size distribution, with 75% of the population having a length between 50 and 250 nm and a width of around 50 nm. Size analysis by dynamic light scattering yields consistent results. Nanocapsules
of smaller size and with a narrower size distribution have been obtained by highpressure extrusion through polycarbonate filters with a 200 nm pore size [9].
3. Delivery of contents
The cytotoxicity of the cisplatin nanocapsules towards the human ovarian carcinoma IGROV-1 cell line has been compared to that of free cisplatin. The IC50 value (the drug concentration at which cell growth is inhibited by 50%) of cisplatin administered as nanocapsules is two orders of magnitude smaller than that of the free drug [9]. The higher cytotoxicity is explained by the reduced inactivation of the drug, due to the lipid coat sequestering it from reaction with substrates in the extracellular environment. Upon binding to the cell surface or endocytic uptake of the nanocapsules, the coat is destabilized, and after membrane passage, cisplatin can exert its cytotoxic effect.
Comments
Cisplatin nanocapsules represent a new lipid formulation of cisplatin, distinct from conventional liposomal formulations in that the drug is present as a nanoprecipitate surrounded by a bilayer. This results in a drug-to-lipid ratio that exceeds that of liposomal formulations by two to three orders of magnitude and probably accounts for the typical bean-like shape of the nanocapsules. The high encapsulation efficiency of cisplatin in nanocapsules is expected to increase the bioavailability of the drug and thus improve the therapeutic index as compared to liposomal formulations of cisplatin. Data obtained so far indicate that the cisplatin nanocapsules can gain access to the cell interior via endocytosis [9]. The method for preparing cisplatin nanocapsules may be applicable to a variety of other compounds with limited solubility in
PROTOCOL 6.13 |
253 |
water and low lipophilicity that are not efficiently encapsulated in conventional liposomes. Alternatively, compounds of interest may be co-encapsulated together with cisplatin.
Like the surface of conventional liposomes, the membrane surface of nanocapsules can be engineered to include poly (ethyleneglycol)-conjugated lipids (reviewed in ref. 2). This results in more stable nanocapsules without affecting the cytotoxicity [12]. It is expected that the technologies developed for liposomes, including the attachment of ligands or antibodies for purposes of targeting, can also be applied to nanocapsules.
Acknowledgement
Financial support by the Dutch Cancer Society (project UU2001-2493) is gratefully acknowledged.
References
1.Drummond, D. C., Meyer, O., Hong, K., Kirpotin, D. B. and Papahadjopoulos, D. (1999)
Pharmacol. Rev. 51, 691–743.
2.Lian, T. and Ho, R. J. Y. (2001) J. Pharm. Sci., 90, 667–680.
3.Garrett, F. E., Goel, S., Yasul, J. and Koch, R. A. (1999) Biochim. Biophys. Acta, 1417, 77–88.
4.Duzg¨unes,¨ N. and Nir, S. (1999) Adv. Drug Deliv. Rev., 40, 3–18.
5.Lippert, B. (1999) Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug. Wiley-VCH, New York.
6.Hacker, M. P. (1991) In: The Toxicity of Anticancer Drugs (G. Powis and M. P. Hacker, eds), p. 82. McGraw-Hill, New York.
7. Newman, M. S., Colbern, G. T., Working, P. K., Engbers, C. and Amantea, M. A. (1999)
Cancer Chemother. Pharmacol., 43, 1–7.
8.Meerum Terwogt, J. M., Groenewegen, G., Pluim, D., Maliepaard, M., Tibben, M. M., Huisman, A., ten Bokkel Huinink, W. W., Schot, M., Welbank, H., Voest, E. E., Beijnen, J. H. and Schellens, J. H. M. (2002)
254 |
IN VITRO TECHNIQUES |
|
|||
|
Cancer |
Chemother. |
Pharmacol., |
49, |
|
|
201–210. |
|
|
|
|
9. |
Burger, K. N. J., |
Staffhorst, R. W. H. M., |
|||
|
de Vijlder, H. C., |
Velinova, M. J., Bomans, |
|||
|
P. H., |
Frederik, P. M. |
and de Kruijff, B. |
||
(2002) Nat. Med., 8, 81–84.
10.Rouser, G., Fleischer, S. and Yamamoto, A. (1970) Lipids, 5, 494–496.
11.Burger, K. N. J., Staffhorst, R. W. H. M. and de Kruijff, B. (1999) Biochim. Biophys. Acta,
1419, 43–54.
12.Velinova, M. J., Staffhorst, R. W. H. M., Mulder, W. J. M., Dries, A. S., Jansen, B. A.
J., de Kruijff, B. and de Kroon, A. I. P. M. (2004) Biochim. Biophys. Acta, 1663, 135–142.
Note added in proof: The molecular architecture of the cisplatin nanocapsules was recently solved (Chupin, V., de Kroon, A. I. P. M., and de Kruijff, B. (2004) J. Am. Chem. Soc., 126, 13816–13821.)

PROTOCOL 6.14
A rapid screen for determination of the protective role of antioxidant proteins in yeast
Luis Eduardo Soares Netto
Introduction
Yeast has been successfully employed as a model to study several biological processes. Indeed, Leland H. Hartwell and Paul M. Nurse received the Nobel Prize in 2001 for the discovery of genes in yeast involved in cell cycle regulation, which has had a great impact on the understanding of processes such as cancer. Some of the advantages of using yeast as a model system are: (1) this micro-organism grows very rapidly and media used to cultivate it are very simple and inexpensive; (2) it is very amenable to genetic manipulations such as gene disruption; (3) its biology is very well studied and (4) its genome has been completely sequenced and is publicly available.
There has been considerable interest in the study of antioxidant enzymes in yeast. Yeast as well as bacteria, mammals and plants possess several enzymatic systems capable of decomposing peroxides.
Saccharomyces cerevisiae possesses five peroxiredoxins (a type of thiol-dependent peroxidase, many of them thioredoxindependent peroxidases); two catalases; three glutathione-dependent peroxidases and one cytochrome c peroxidase. Although a partial redundancy of their roles is observed, they definitively have distinct roles. For example:
cytosolic thioredoxin peroxidase I (cTPxI) is important for protection of cells with dysfunctional mitochondria, whereas mitochondrial thioredoxin peroxidase I is important for respiratory-competent cells [1– 3]. More recently, results have been obtained indicating that cytosolic thioredoxin peroxidase II is specifically important for cell protection against organic peroxides independently of the functional state of mitochondria [4]. One approach that has been very helpful for us is a viability test based on serial dilutions as described below. Using this approach, we were able to test mutant strains under several different conditions and found one in particular, where a gene deletion rendered cells very sensitive to oxidative stress. Several concentrations of oxidants can be tested as well as the effect of various carbon sources. Changing the carbohydrate present in the media is an interesting manipulation because yeast physiology and biochemistry are very dependent on the carbon source. For example, yeast grown under glucose get most of its ATP from glycolysis, has few mitochondria and this organelle contains few cristae. In contrast, when yeast is grown in glycerol or ethanol it only produces ATP from oxidative phosphorylation and possesses many active mitochondria [5].
It is important to emphasize that this approach can also be employed for the

256 IN VITRO TECHNIQUES
study of other protective roles, for example against heat stress, osmotic stress and alkylation damage. Proteins from other organisms can also be studied. In this case, it would be necessary to verify if a heterologous gene can complement the sensitivity observed for a given mutant strain.
After testing several conditions by this protocol and finding one where the mutant strain is specifically sensitive, a more accurate and quantitative assay (colony counting) can be performed. A disadvantage of colony counting assay is that it is very time consuming. Examples of this strategy are described in refs [2] and [4].
Reagents
Preparation of medium and plates
A. Synthetic medium
Per litre, add:
1.7 g Yeast nitrogen base without amino acids or ammonium sulfate (YNBAA/AS)
5 g (NH4)2SO4
20 g of glucose or other carbon source
(2%)
This medium can support the growth of yeast with no nutritional requirements. Generally, however, yeast strains present nutritional requirements. Therefore, nutrients should be added as described in Table 6.1. Synthetic media should be autoclaved before use. Threonine and aspartic acid work better when added after autoclaving, therefore stock solutions of these amino acids should be prepared and sterilized by filtration. These amino acids can then be added to a synthetic medium in laminar flow.
B. Preparation of plates
Solid synthetic medium is prepared as described above with addition of agar
Table 6.1 Nutrients to be added with yeast strains with deficiencies in metabolic processesa
Nutrient |
Final |
Amount |
|
concentration |
of nutrient |
|
(ug/ml) |
added (mg)b |
Adenine |
40 |
40 |
(hemisulfate |
|
|
salt) |
|
|
L-arginine (HCl) |
20 |
240 |
L-aspartic acid |
100 |
100 |
L-glutamic acid |
100 |
100 |
(monosodium |
|
|
salt) |
|
|
L-histidine |
20 |
20 |
L-leucine |
60 |
60 |
L-lysine |
30 |
30 |
(mono-HCl) |
|
|
L-methionine |
20 |
20 |
L-phenylalanine |
50 |
50 |
L-serine |
375 |
375 |
L-threonine |
200 |
200 |
L-tryptophan |
40 |
40 |
L-tyrosine |
30 |
30 |
L-valine |
150 |
150 |
Uracil |
20 |
20 |
aThese data were obtained from Ausubel et al. [6].
bThe amount of nutrient was added per litre of synthetic media.
2% (20 g of agar per litre of medium). The medium is autoclaved and then left for 45–60 min at room temperature until cooled to about 50 ◦C, which can be felt if one can hold the flask comfortably. At this point, the medium should be poured into plates. Drugs and other compounds to be tested should also be added when the medium is at 50 ◦C, to avoid undesirable thermal decomposition. More details about preparation of media can be obtained from Ausubel et al. [6].
Procedure
Determination of hydroperoxide tolerance
1.Grow yeast cells overnight in complete synthetic media with various carbon
PROTOCOL 6.14 |
257 |
sources; in most cases, 2% glucose or with 2% glycerol.
2. The next day, dilute inoculate to 3 ×
106 cells per ml (which corresponds to approximately 0.1 OD600 nm) and then cultivate until the cell density reaches approximately 3 × 107 cells per ml (which corresponds to approximately 1.0 OD600 nm). This takes about 10 h, which ensures that cells are well adapted to the carbon source present in the medium.
3.Dilute cell suspension to OD600 nm = 0.2 and make four further 1/5 dilutions of the cell suspensions.
4.Place a 10 µl droplet of each dilution into complete synthetic medium + agar with 2% glucose or the other carbon sources as described in Figure 6.10. A
OD600 nm = 0.2
control plate should contain media components alone, whereas the test plate contains peroxide (or other compound to be investigated).
5.Incubate plates for 30 or 48 h, depending on the results observed. In some cases, a differential effect can only be found after only one of these time periods.
Comments
1.In the control plate, there is no addition besides the components of the media. It is important to be very careful in
the determination of OD600 nm before the dilutions in order to guarantee that mutant and wild-type strains will grow equally.
2.Cells are grown in synthetic medium to avoid the reaction of peroxides with
WT |
1/5 |
1/5 |
|
1/5 |
1/5 |
|
|
|
|
|
|
|
|
Dilution |
0 |
1/5 |
1/25 |
1/125 |
|
1/625 |
10 ul |
10 ul 10 ul |
|
|
10 ul |
10 ul |
|
|
|
10 ul |
|
10 ul 10 ul |
|
|
|
|
10 ul |
|
|
||
|
|
10 ul |
|
media + |
||
control |
|
|
|
|
||
|
|
|
|
|
peroxide |
|
plate |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 ul 10 ul |
|
10 ul |
|
|
|
|
|
10 ul |
|
10 ul |
|
|
10 ul 10 ul |
|
|
|
||
|
|
|
10 ul 10 ul |
|
||
10 ul |
|
|
|
|
|
|
Dilution |
0 |
1/5 |
1/25 |
1/125 |
|
1/625 |
mutant |
1/5 |
1/5 |
|
1/5 |
1/5 |
|
|
|
|
|
|
|
|
OD600 nm = 0.2
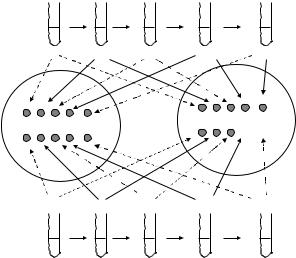
Figure 6.10 Scheme viability of yeast cells in plates after serial dilutions. Cells were submitted to four 1/5 dilutions (200 ul to 1 ml) in synthetic media; 10 µl of undiluted and 1/5, 1/25, 1/125 and 1/625 diluted suspensions were added to control plates as well as to the plates with the compounds to be tested. In control plates, the two strains grew equally which showed that their cell densities were the same in the undiluted samples. In the experimental plate, the mutant strain did not grow in the 1/125 and in the 1/625 dilutions, indicating that this gene deletion rendered cells sensitive to peroxide treatment
258 IN VITRO TECHNIQUES
extracellular components present in a rich medium such as glutathione [7]. It is important to use synthetic rather than rich media such as YPD (yeast peptone dextrose) because glutathione and other substances are not very well controlled and their concentration can vary from different lots of media, which may compromise the reproducibility of the assay.
3.Peroxides, mitochondrial inhibitors or other stressful compounds can be added to the plates under several different conditions, in order to find one where deletion of a gene severely retards growth. Plates can be incubated for different intervals, which can also interfere with the results. We usually incubate plates for 30 or 48 h. It may be interesting to vary the carbon source because it provokes dramatic changes in yeast biochemistry and physiology as mentioned above.
4.It is important not to use small volumes to make the dilutions. We generally diluted 200 µl of cell suspension into 1 ml of synthetic media. In this way, errors due to manipulation of pipettes are minimized.
References
1. Kowaltowski, A. J., Vercesi, A. E., Rhee, S. G. and Netto, L. E. S. (2000) Catalases and
thioredoxin peroxidase protect Saccharomyces cerevisiae against Ca2+-induced mitochondrial membrane permeabilization and cell death. FEBS Lett., 473, 177–182.
2.Demasi, A. P. D., Pereira, G. A. G. and Netto, L. E. S. (2001). Cytosolic thioredoxin peroxidase I is essential for the antioxidant defense of yeast with dysfunctional mitochondria. FEBS
Lett., 509, 430–434.
3. Monteiro, G., Pereira, G. A. G. and Netto, L. E. S. (2002) Regulation of mitochondrial thioredoxin peroxidase I expression by two different pathways: one dependent on CAMP and the other on heme. Free Radical Biol. Med., 32, 278–288.
4.Munhoz, D. C. and Netto, L. E. S. (2004) “Cytosolic thioredoxin peroxidase I and II are important defenses of yeast against organic hydroperoxide insult, J. Biol. Chem., 279, 35219–35227.
5.Pon, L. and Schatz, G. (1991) Biogenesis of Yeast Mitochondria in the Molecular and Cellular Biology of the Yeast Saccharomyces. Vol 1: Genome Dynamics, Protein Synthesis and Energetics. Cold Spring Harbor Laboratory Press, New York.
6.Ausubel, F. M., Brent, R., Kingstone, R. E., Moore, D. D., Seidman, J. A., Smith, J. A. and Struhl, K. (1994) Saccharomyces cerevisiae. In: Current Protocols in Molecular Biology, John Wiley & Sons, Inc., New York.
7.Maris, A. F., Assump¸cao,˜ A. L., Bonatto, D., Brendel, M. and Henriques J. A. (2001) Diauxic shift-induced stress resistance against hydroperoxides in Saccharomyces cerevisiae is not an adaptive stress response and does not depend on functional mitochondria. Curr. Genet., 39, 137–149.

PROTOCOL 6.15
In vitro assessment of neuronal apoptosis
Eric Bertrand
Introduction
Apoptosis is a specific form of cell death which is triggered and executed by the cell’s own machinery; as such it is considered as a kind of ‘suicide’. Elimination of cells through apoptosis contributes to many important physiological processes such as development, inflammation and adult tissue homeostasis [1, 2]. Also, excessive apoptosis has been associated with a number of conditions, like neurodegenerative diseases, ischaemia or Aids. At first, the definition of apoptosis was essentially based on morphological features: cell shrinkage, condensation of cytoplasm and nucleus, clumping of chromatin, blebbing of the membrane and ultimately fragmentation in apoptotic bodies. Afterwards, it was observed that the alterations of chromatin were also accompanied by a cleavage of nuclear DNA into nucleosomal fragments; however, this type of DNA degradation is not entirely specific of apoptosis. At the biochemical level, most of the characteristic changes observed during apoptosis are mediated by the caspases, a family of cysteine proteases. The caspases cleave a number of substrates within the cell including structural proteins and enzymes, thereby modifying their function. Caspase activation is currently considered as the most reliable marker of apoptosis.
The protocols included therein should prove useful to the reader who wants
to investigate whether a particular treatment induces apoptosis on neural cell cultures. Some of the issues that should be addressed in the course of such a study are: is the treatment toxic, does it induce apoptosis, which caspases are activated? Toxicity can be assessed and quantified with the TUNEL assay, while reversal of the toxic effect by a caspase inhibitor such as ZVAD indicates the involvement of apoptosis. ZVAD is a broad-range caspase inhibitor, some caspase inhibitors are more selective than ZVAD, but they are usually not entirely specific. For instance, DEVD preferentially inhibits caspase-3 but is also active against caspase 8, 7 and 10 [3]. Therefore, even if DEVD blocks the apoptosis induced by the treatment, a direct demonstration of caspase-3 activation using immunostaining is required. Finally, a very specific inhibition of a given caspase can be achieved with antisense oligonucleotides coupled to the cell permeable carrier penetratin.
Reagents
PBS: GIBCO/Invitrogen, cat. no. 14200059
zDEVD-fmk: Tebu, cat. no. P414
zVAD-fmk: France Biochem, cat. no. 727610
Vectastain ABC-AP kit: Vector Laboratories, cat. no. AK-5200
NBT/BCIP: Pierce, cat. no. 34042
260 IN VITRO TECHNIQUES
In situ Cell Detection Kit, AP: Roche Diagnostics Cat N◦ 1 684 809
Anti p20 antibody: Pharmingen, cat. no. 557035
J peptide: Syntem,
Penetratin peptide and oligonucleotides: Quantum, cat. no. 151300
Procedures
Immunocytochemical methods to study apoptosis at the single cell level
The protocols within this section are intended for cells that have been cultured in ELISA wells; with some adjustments on solution volumes they can be adapted to glass coverslips and other plate formats. The cells were fixed with 4% paraformaldehyde at room temperature for 30 min or overnight at 4 ◦C.
TUNEL staining
TUNEL stands for TdT mediated dUTP nick end labelling; this reaction labels DNA strand breaks by incorporating modified nucleotides at the 3 -OH termini. During apoptosis, nuclear DNA is degraded to nucleosomal fragments which are detected by the TUNEL reaction. In this protocol we used the alkaline phosphatase version of the In Situ Cell Death Detection Kit provided by Roche Diagnostics. In most cases we followed the instructions of the manufacturer, however we obtained a better specificity of the staining on primary cultures of cortical neurons by reducing the incubation times for the 1 + 2 reaction mixture (DNA labelling solution) and the converter-AP solution (alkaline phosphatase coupled antibody). Some adjustment of incubation times and other parameters might be necessary, depending on
the cell type studied and the type of signal observed (fluorescence, alkaline phosphatase, horseradish peroxydase); the manufacturer gives a number of useful pieces of advice in this respect (http://www.roche- applied-science.com). This protocol has been used to detect apoptosis on rat and mouse cortical neurons and it should be applicable to other species and neuronal types.
1.Wash cells 3× with PBS and then permeabilize in a 0.1% sodium cit-
rate/0.1% triton solution for 2 min on ice (at 4 ◦C).
2.Wash wells 2× with PBS.
3. Add 50 µl of TUNEL 1 + 2 reaction mixture (enzyme solution + label solution) from Roche Diagnostics kit for 30 min at 37 ◦C.
4.Wash wells 3× with PBS.
5.Incubate the cells for 15 min at 37 ◦C in 50 µl of converter-AP solution (alkaline phosphatase coupled antibody).
6.Prepare NBT/BCIP substrate solution for alkaline phosphatase:
100mM Tris pH 9.5
10mM NaCl
5mM MgCl2
1mM levamisole to inhibit endogenous phosphatases
7.Add NBT/BCIP according to the manufacturer’s guidelines.
8.Wash wells 3× in PBS, add 100 µl of NBT/BCIP substrate solution and incubate for approximately 10 min at room temperature. Follow the course of the reaction and block it by adding
10µl of 0.5 M EDTA in the wells when the staining is intense enough.