

Основные параметры процесса.
1. Температура. С повышением Т скорость реакции изомеризации возрастает до предела, ограничиваемого равновесием процесса. Дальнейшее повышение t приводит к усилению реакции гидрокрекинга с образованием газообразных продуктов, при этом возрастает расход водорода, а выход изомеров снижается.
2. Давление. Р оказывает влияние на равновесие реакции изомеризации н-парафинов, но Р существенно влияет на кинетику целевых и побочных реакции процесса. Т.о повышение Р при прочих равных условиях снижает глубину, но повышает селективность изомеризации. Увеличение парциального давления водорода снижает скорость дезактивации кат., т.к. тормозится коксообразование. Повышение Р выше 4МПа не целесообразно, т.к. коксобразование практически не меняется.
3. Объемная скорость подачи сырья (w). При постоянной степени превращения w и t оказывают противоположное влияние на скорость изомеризации. Для увеличения w в двое требуется повышение t процесса на 8-11’С.
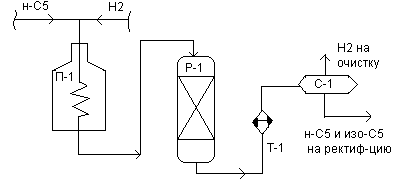
Установка изомеризации фракции Н.К.-62’С. Изомеризацию на соответствующих промышленных установках проводят с одновременной ректификацией реакционной смеси и циркуляцией не превращенного сырья. Это связано с тем, что степень превращения сырья на кат ИП-62 около 52%. Исходное сырье предварительно подвергают гидроочистке и осушке. В блоке ректификации производится выделение изомеров из смеси исходного сырья и стабильного изомеризата. Реакторный блок состоит из 2-х параллельно работающих секций, в одной проводится изомеризация н-пентанов, а в другой н-гексанов.
Пентановая фракция, выделяемая в блоке ректификации и содержащая ~ 91% н-гептана смешивается с ВСГ, нагревается в трубчатой печи П-1 до требуемой Т и идет в реактор изомеризации со стационарным слоем кат-ра Р-1.Парогазовая смесь продуктов реакции охлаждается и конденсируется в тепл-ках и холод-ках и идет в сепаратор С-1. Циркулирующий ВСГ из сепаратора после осушки в адсорбере подается компрессором на смешение с сырьем. Изомеризат после С-1 смешивается с сырьем и подается на ректификацию. Аналогична схема и для гексановой фракции. Основные параметры и показатели работы установки: Т=360-430; Р=3,2-3,6 МПа; Н2: сырьё = (2-3) : 1; w = 2-2.3 ч-1; ОЧИМ изопентана = 89,5 ; Срок службы кат-ра 13-46 месяцев; Содержание изопентана в готовой продукции 96,5-99,5 %. Расход Н2 в процессе изомеризации невелик 0,1-0,3% на сырьё. Себестоимость изомеризатов в 3 раза ниже, чем алкилатов. При этом процесс изомеризации имеет более надежную сырьевую базу, чем алкилирование.
В-21.Каталитическое гидрооблагораживание нефтяного сырья (гидроочистка). имзм и параметры процесса, технологическая схема установки гидроочистки ДТ.
Цели процесса:
1). Гидроочистка моторных топлив от S, O2, N2, As, галогенов Ме и гидрирование непредельных УВ. Это уменьшает коррозионную агрессивность топлив, их склонность к образованию осадков, уменьшить выброс токсичных газов в атмосферу.
2). Для защиты платиновых кат-ров риформинга от отравления не УВ-ми соединениями.
3). Для повышения выхода и качества продуктов крекинга гидроочистке подвергают вакуумные газойли.
4). Для осветления, снижения коксуемости, кислотности и эмульгируемости нефтяных масел.
Химизм:
При гидрогенолизе происходит разрыв связей C-S, C-N, C-O и насыщение водородом образующихся гетероатомов и двойных связей. При этом S, O2, N2 выделяется в виде H2S, NH3, H2O. RSH + H2→ RH + H2S ; RSR + H2→ RSH + RH; Циклические сульфиды гидрируются с образованием олифатических УВ и H2S. Азот в нефтяном сырье находится в гетероциклах в виде производных пиррола и пиридина. Кислород в нефтяных фракциях находится в виде спиртов, эфиров, фенолов и кислот. В тяжелых нефт.остатках O2 находится в основном в мостиковых связях и в циклах полициклических ароматических и АСВ нефти.
Реакции гидрогенолиза экзотермичны, протекают либо без изменения, либо с уменьшением объема Для серосодержащих соединений скорость гидрогенолиза изменяется в следующем ряду : RSH >RSSR’>RSR’>C4H8S>C4H4S
Катализаторы процессов гидроочистки нефтяных фракций.
1) Металлы 8 группы: Ni, Co, Pt, Pd, Fe.
2)Окислы и сульфиды: Mo, W, Cr, MoS3
Кроме названных кат-ров имеются носители с развитой удельной поверхностью, высокой механической прочностью. Они инертные или с кислотными свойствами.
Ni, Co, Pt, Pd – кат-ры гидрирующего и дегидрирующего действия. Они не обладают устойчивостью к действию контактных ядов и не могут использоваться отдельно в процессах гидроочистки. Наибольшее распространение в гидрогенизирующих процессах получили следующие катализаторы: АКМ, АНМ, АНКМ, АНМС, АНВ, АКВ. Получают развитие цеолитсодержащие катализаторы. Предпологается, что гидрогенолиз гетероорганических соединений протекает многостадийно через хемосорбцию участников реакции на активных центрах Ni или Co, Mo или W. При этом на Co и Ni происходит активация водорода и спилловер атомарного активного водорода. Параметры процессов гидрогенизации.
1)Сырьё- бензиновые, керосиновые и дизельные фракции, а также вакуумный газойль и смазочные масла, содержащие S, N и непредельные УВ. По мере утяжеления сырья увеличивается не только общее содержание, но и доля наиболее устойчивых к гидрогенолизу гетероатомных соединений.
2) Температура, объемная скорость сырья и давление- они определяются кинетикой процесса. Так для ДТ требуемая глубина обессеривания 90-93% достигается при объемной скорости 4 ч-1, давлении 4 МПа и 350-380ºС. При Т=420ºС из-за реакций гидрокрекинга возрастает выход газов и легких УВ, увеличивается коксообразование и расход водорода..
3) Парциальное давление водорода и краткость циркуляции ВСГ. При повышении общего давления растет Рпарц водорода. На Рпарц водорода влияет кратность циркуляции ВСГ и концентрация водорода в ВСГ. Чем выше содержание водорода, тем ниже кратность циркуляции ВСГ. Кратность циркуляции возрастает с утяжелением сырья. Процессы гидроочистки проводят в адиабатических реакторах без отвода тепла реакции. При гидроочистке высококипящих и высокосернистых фракций предусматривается отвод тепла за счет подачи холодного ВСГ между слоями ВСГ.
4) Регенерация кат-ра. Кат-р теряет свою активность за счет закоксовывания и отложения на его поверхности тяжелых металлов, регенерацию проводят паро-воздушной смесью при Т= 530ºС.
Принципиальная тех. схема уст-ки гидроочистки ДТ ЛЧ-24-2000.
Циркуляционный ВСГ смешивается с сырьем, смесь нагревается в сырьевых теплообменниках и в трубчатой печи П-1 до Т реакции и поступает в реактор Р-1. После реактора газо-продуктовая смесь частично охлаждается в сырьевых теплообменниках до Т= 210-230ºС и поступает на сепарацию ВСГ в сепараторы С-1 и С-2. ВСГ после С-2 поступает на очистку МЭА в абсорбер К-2 и далее на смешение со свежим ВСГ в качестве циркуляционного газа. Гидрогенезаты горячего из С-1 и холодного из С-2 сепараторов смешиваются и направляются в стабилизационную колонну К-1, где подачей подогретого в печи П-1 отдувочного ВСГ из гидроочищенного продукта удаляются УВ-ные газы и бензин. Расход водорода составляет 0,4% масс. на сырье. Выход гидроочищенного топлива 96,9%, бензина–1,3%, УВ-ных газов–0,6%, сероводорода–1,2% и потери 0,4%.
Параметры уст-ки ЛЧ24-2000:
Мощность 2 млн. т/год по сырью. Расход Н2 0,4% масс
Р=5 МПа. W=4,5 ч-1. Т=360-400 ºС.
В-22.Каталитический риформинг. Основы процесса. Технологическая схема.
Кат.риформинг предназначен для повышения детонационной стойкости бензинов, получения индивидуальных аренов (бензола, толуола, ксилолов), для получения дешевого водородсодержащего газа.
Химизм и термодинамика процесса. Целевыми в каталитическом риформинге являются реакции образования ароматических углеводородов за счет: 1.дегидрирование шестичленных цикланов;
2.дегидроизомеризации циклопентана;
2. дегидроциклизации парафиновых УВ (С6 - С7)
Процессы параллельно протекают с нежелательными реакциями гидрокрекинга с образованием как низко так и высококипящих УВ.
Катализаторы и механизм их действия.
Кат.риформинг осуществляется на бифункциональных кат-рах, состоящих из металлов и оксидов металлов. Основным компонентом катализатора являются металлы: Pt промотированная добавками рения и иридия, олова, галия и германия, тонко диспергированные на носителе. Вторым компонентом катализатора является оксид алюминия Al2O3, одновременно является носителем для Pt и ее промоторов. Для усиления и регулирования кислотной функции носителя в состав кат-ра вводят галогены: F или Сl. Платина на кат-ре не только ускоряет реакции гидрирования и дегидрирования, но и замедляет образование кокса на его поверхности.
Параметры процесса.
1). Качество сырья риформинга - определяется фракционным и химическим составом бензина. Качество сырья влияет на выход риформата, октановое число (выход аренов), выход водорода, закоксовывание катализатора. С увеличением молекулярной массы сырья выход реформата возрастает, аналогичным образом изменяется выход аренов и ОЧ. Состав сырья и его фракционный состав влияют на закоксовывание кат-ра, Увеличение числа атомов углерода в аренах и нафтенах сырья более 7 - 9 приводит к повышенному закоксовыванию кат-ра. 2). Температурный режим процесса и распределение объема катализатора по реакторам. Т.к. процесс риформирования сильно эндотермичен его проводят в каскаде из 3 - 4 реакторов с промежуточным подогревом сырья. В первом реакторе протекает высоко эндотермическая реакция дегидрирования нафтенов, в последнем реакторе протекают преимущественно эндотермические реакции дегидроциклизации и экзотермические реакции гидрокрекинга парафинов. В первом реакторе наибольший перепад Т=30-50ºС, в последнем реакторе наименьший перепад Т. Высокий температурный градиент в первом реакторе понижают ограничивая в нем глубину реакций ароматизации нафтенов. Это достигают уменьшением объема кат-ра в нем, а следовательно уменьшением времени контакта сырья. В этой связи на промышленных установках головной реактор имеет наименьший объем кат-ра, а конечный наибольший. По мере закоксовывания кат-ра и потери его активности Т на входе в первый реактор постепенно повышают, обеспечивая заданное качество катализата. Среднее повышение Т на входе в реактор 0,5-2ºС в месяц. Максимальная Т сырья на входе в последний реактор достигает 535ºС.
3). Давление. Оно оказывает влияние на выход и качество риформата. при прочих равных параметрах с понижением парциального давления водорода возрастает глубина ароматизации сырья как с термодинамической, так и кинетической точки зрения, при этом повышается селективность превращения парафинов и тормозит реакции гидрокрекинга. Однако при снижении давления возрастает скорость дезактивации кат-ра за счет его закоксовывания. Скорость дезактивации кат-ра обратнопропорциональна давлению.
4). Кратность циркуляции ВСГ. Определяется как отношение объема циркулирующего ВСГ при нормальных условиях к объему сырья в единицу времени. Рекомендуемый расход водорода 900 - 1500 м3 ВСГ / м3 сырья. С увеличением мольного соотношения водород - сырье скорость дезактивации катализатора сокращается, а межрегенерационный цикл удлиняется. Однако увеличение Квсг приводит к увеличению энергозатрат, росту гидравлических сопротивлений и объема аппаратов. 5). Объемная скорость подачи сырья (W)- оказывает влияние на риформинг как параметр, обратный времени контакта сырья с кат-ром. С увеличением W снижается глубина реакции ароматизации и более значительно реакций гидрокрекинга парафинов. В результате повышение W приводит к:
1)к увеличению выхода риформата, но с пониженным октановым числом и меньшим содержанием аренов;
2)снижению выхода ВСГ, но с более высокой концентрацией водорода; 3)повышению селективности процесса и удлинению межрегенерационного цикла. Обычно W в процессах риформинга составляет 1,5 - 2 ч-1.
6). Содержание Cl в катализаторе. Cl обеспечивает активность кислотных центров кат-ра, но т.к. Сl летучий, то кислотность центров понижается. Потери Cl восполняются постоянной подачей Cl в сырье. При регенерации кат-ра Cl в виде ССl4 или C2H4Cl2 подается в течении 2 - 10 часов при 500 - 520ºС в количестве 0,5 - 1,5 % от массы катализатора.
Тех. схема. Гидроочищенное и осушенное сырье смешивается с циркулирующим ВСГ, подогревается в теплообменнике, затем в секции печи П-1 и поступает в реактор первой ступени Р-1. На установке имеются 3-4 адиабатических реактора и соответствующее число секций в печи П-1 для межступенчатого подогрева реакционной смеси. На выходе из последнего реактора смесь охлаждается в теплообменнике и холодильнике до 20-40ºС и поступает в сепаратор высокого давления С-1 для отделения циркулирующего ВСГ от катализата. Часть ВСГ после осушки цеолитами в адсорбере Р-4 поступает на прием циркуляционного компрессора, а балансовый избыток отводится в блок гидроочистки бензола и передается другим потребителям водорода. Нестабильный катализат из С-1 поступает в сепаратор низкого давления С-2, где от него отделяют легкие УВ. После С-2 газовая и жидкая фазы поступают в абсорбер К-1. Абсорбентом служит стабильный катализат– бензин. Низ абсорбера подогревается горячей струей через печь П-2. В абсорбере при P=1,4 МПа и Тниза=165 и Тверха=40ºС отделяется сухой газ (IV). Нестабильный катализат с низа К-1 после подогрева в теплообменнике поступает в колонну стабилизации К-2. Тепло в низ К-2 подводится за счет циркуляции стабильного катализата через печь П-2. Головная фракция стабилизации после конденсации и охлаждения поступает в приемник С-3, откуда частично идет на орошение К-2. Балансовый избыток выводится с установки. Часть стабильного катализата с низа К-2 после охлаждения в теплообменнике подается во фракционирующий абсорбер К-1. Балансовый избыток выводится с установки.
В-23. Сущность реакций алкилирования их класс-ция. алкилирующие агенты и катализаторы. Энергетическая характеристика основных реакций алкилирования.
Алкилированием наз-ют процесс введения алкильных групп в молекулы органических и нек-ых неорганических веществ. Эти процессы имеют большое практическое значение прежде всего для синтеза алкилароматических УВ, к-ые исп-ют в производстве синтетических каучуков, ПАВов, пластмасс, синт.волокон. Ведущее место занимает алкил-ие бензола олефинами. В больших масштабах производят продукты алкилирования меркаптанов, парафинов, сульфидов, аминов, спиртов.
Классификация процессов алк-ния:
1) По атому углерода (С-алкилирование). Такое замещение происходит в парафинах и аренах. Kat
ArH + RCl → ArR+HCl
AlCl3
2) По атомам кислорода и серы (O- и S- алк.).
ArOH + RCl +NaOH→ ArOR+NaCl+H2O
NaSH + RCl → RSH + NaCl
3)По атому азота (N-алк). В аммиаке и аминах Н2 замещается на Алк.
NH3 + RCl → RNH2 + HCl
4) По атомам других элементов (Si,Pb,Al). это важнейший путь получения металлорганических соединений.
2RCl + Si→R2SiCl2
Реа-ции алк-ния различают по строению алкильной группы, вводимой в соединение:
- циклоалкилирование циклогексилбензол
![]()
-арилирование С6H5Cl +NH3 → C6H5NH2 + HCl
- винилирование ROH + CH≡CH → ROCH═CH2
Алкильные группы могут содержать различные заместители – Cl, -COOH, -NO2,-OH.
Например, β- оксиалкилирование C2H4-O +HOH→ HO-CH2-CH2-OH.
Алкилирующие агенты и катализаторы.
1) Олефины. Чаще прим.этилен, пропилен, бутилен и высшие жидкие олефины. Их прим. в основном для С-алк-ния парафинов и аренов. Они не прим-ся для N-алк-ния и не всегда эффективны при S-и О- алк. Алкилирование при этом протекает по ионному механизму и катализируется протонными и апротонными кислотами.
R-CH=CH2 +H+→ R-C+H-CH3, карб-катион
В соответствии с устойчивостью карб-катиона реакционная способность олефинов изменяется в ряду:
CH2=C(CH3)2 > i-CH2=CH-CH2-CH3 > CH3-CH=CH2 > CH2=CH2
Удлинение и разветвление цепи углеродных атомов в олефине значительно повышает его реакционную способность.
2) Алкилгалогениды, в основном прим.хлорпроизводные. Они пригодны для всех типов реакций алк-ния. Механизм электрофильный и нуклеофильный, иногда радикальный. Для С-алкилирования характерен мех-зм электрофильного замещения в присутствии апротонных кислот AlCl3, FeCl3:
RCl + AlCl3 → R+ + AlCl4+
Реакционная способность зависит от устойчивости карб-катиона и повышается при удлинении и разветвлении алкильной группы:
(CH3)3 –C-Cl > (CH3)2 –CH-Cl >CH3 –CH2-Cl
При N-,S-,O-алк.механизм реакции нуклеофильный и без катализатора:
RCl + :NH3 →[ Cl δ----R---NH3δ+] → Cl - + RNH2+ H+
Реакционная способность алкилгалогенидов в реаях нуклеофильного замещения изменяется в ряду:
ArCH2Cl > CH2=CH-CH2Cl > AlkCl > ArCl; Перв. AlkCl > втор.AlkCl > трет.AlkCl
3) Спирты и простые эфиры. Спирты прим.для реакций O-,N-алк. В тех случаях когда они дешевле и доступнее хлорпроизводных. Процесс идет под действием кислотных Kat.
..
R-:O:H + H+ ↔ R-O δ+-H→ H2O + R+
H
Из простых эфиров исп. только оксиды олефинов. Они исп.для O-,N,-,S-алкил. Реакции идут под действием основных кат-ров или без катал-ра.
Энергетическая характеристика основных реакций алкилирования.
Все р-ции алкирирования экзотермичны, но значения тепловых эффектов сильно различаются в зависимости от типа разрывающейся связи и алкилирующего агента. При одном и том же алкилирующем агенте ∆Н реакции уменьшается в след.порядке:
Саром.> Солиф. > N > O
Для разных алкил.агентов существует след.зависимость ∆Н:
СН≡СН > С2Н4О > R-CH=CH2 > ROH > RCl
В-24. Алкилирование ароматических УВ на хлориде алюминия.
Алкил-щие агенты и катализаторы.
В качестве алкилирующих агентов используются хлорпроизводные и олефины. Использование спиртов мение эффективно так как катализатор разлогается образующейся водой.
При реакции с хлорпроизводными образуется сильно поляризованный комплекс или карбкатион. При алкелировании с олефинами добовляют сокотализатор HCl, H2O, RCl.
В случае использования низших олифинов из ацителена образуется первичный алкил бензол , из пропилена вторичный а из изобутелена – третбутил.
Химизм и механизм процесса. Алк-ие бензола олефинами идет через образование карб-катиона. При катализе протонными к-ми он образуется за счет присоединения протона к олефину: СН2=СН2 +Н+ → СН3-С+Н2
nC6H6
Бензол
+ HCl + Al2Cl6
↔
![]() [ *(n-1)C6H6
]+ Al2Cl7-
; n =1-6
[ *(n-1)C6H6
]+ Al2Cl7-
; n =1-6
Этот комплекс очень активный,т.к. легко отдает протон молекуле олефина:
Н+ + R-CH=CH2 → R-CH+-CH3 атакует молекулу олефина.
Бензол
+ R-CH+-CH3
→ [![]() ] →[
] →[
![]() ] → Н+
+
] → Н+
+
![]()
π -комплекс σ- комплекс алкилбензол
При алк-нии высшими олефинами наблюдается изомеризация алк-ных групп, к-ая протекает в направлении образования наиболее устойчивого карб-катиона, но без нарушения углеродного скелета алк-ной группы:
СН3-СН2-СН2-СН=СН2 + Н+ → СН3-СН2-СН2-С+Н-СН3 ↔ СН3-СН2-С+Н-СН2-СН3
Заместители в ароматическом ядре влияют на процесс алк-ния след.образом:
- электроно-донорные заместители (-СН3) увеличивают скорость алк-ния в небольшой степени;
- Хлорбензол алкилируется в 10 раз медленнее бензола.
- Сильные электронно- акцепторные заместители (-СООН, -NO2 -CN) полностью дезактивируют ароматическое ядро, т.е. соединения,сод.эти группы не алкилируются.
В более жестких условиях у гомологов бензола происходит изомеризация с внутримолекулярной миграцией алкильных групп с образованием более устойчивого изомера:
Способность алкильных групп к миграции изменяется в ряду: (СН3)3С > (СН3)2СН > СН2-СН3 >> СН3

Последовательность алк-ния, селективность процесса.
Для повышения селективности процесса необходим 5-10 кратный избыток бензола. Его отгоняют и возвращают в процесс. В прис-вии кат-ров имеет место реакция переалкилирования:
С6Н4(С2Н5)2 + С6Н6↔2 С6Н5-С2Н5
Селективность процесса увеличивается при возвращении получающихся полиалкилбензолов (ПАБ) на реакцию. При этом избыток бензола уменьшают до 2-3 кратного.
На селективность процесса влияют также побочные реакции :
1) Смолообразование
2)Деструкция алк.групп происходит на стадии образования карб-катиона
R-C+H-CH2-R → R-CH=CH2 +R+ +2C6H6→ C6H5-CHR-CH3 + C6H5-R
3) полимеризация происходит в рез-те последовательного взаимодействия карб-кат. с олефинами
СН3-C+H2 + С2Н4→ СН3-СН2-СН2-C+H2 …..
Эти реакции увеличиваются при повышении Т. Их подавляют избытком бензола, и при более низких концентрациях кат-ра ведут процесс.
Технологическая схема получения этил- и изопропилбензола. Состоит из нескольких узлов:
- узел осушки бензола
- приготовления кат-кого комплекса
-проведения реакции алк-ния
-обработка и разделение продуктов реакции.
Свежий бензол вместе с возвратным со стадии разделения идет в к-ну 3,в к-ой происходит осушка бензола азеотропной ректификацией. С верха к-ны выходит низкокипящая азеотропная смесь бензола с водой. Она конденсируется в конд-ре 4 и разделяется в сеп-ре 5 на два слоя. Воду с растворенным в ней бензолом отводят или исп-ют для промывки реакционной массы. Бензольный слой из С-5 стекает на верхнюю тарелку к-ны 3 создавая орошение. Осушенный бензол из куба К-3 подогревается в теплообменнике 2 и поступает в сборник 7,откуданасосом закачивается в алкилатор 9. Каталитический комплекс готовится в аппарате 8 с мешалкой и рубашкой для обогрева паром.В него загружают смесь бензола и ПАБов =1:1 и AlCl3 в количестве 1 моль на 2-3 моля ароматических УВ. После этого при Т и перемешивание подают этилхлорид или небольшое количество воды.
Бензол с этилхлоридом в прис-вии кат-ра образуют этилбензол. А в производстве ИПБ добавляют воду:
AlCl3 +Н2О = AlОНCl2 + НCl
Приготовленный кат-кий комплекс при температуре 60-65 периодически поступает в алкилатор 9(адиаб. колонного типа),где идет реакция алк-ния. В низ 9 поступает газообразный олефин, бензол из 7, рециркулирующий кат.комплекс из С-16 и клнденсат из С-11. Мольное соотношение бензола к олефину =3-3,5:1, Т=100-130, Р=3-4 атм. в алк-ре 9. Парогазовая смесь с верха колонны проходит конд.-холод. 10 где конденсируется бензол. В С-11 конденсат бензола отделяется и возвращается в алкилатор. Газы из С-11 содержат еще много паров бензола. Для его улавливания они идут в абсорбер 12, к-ый орошается ПАБами. Раствор бензола в ПАБах идет в реактор для переалкилирования. Газы после абсорбера 12 промывают щелочью в аб-ре 13 для удаления НCl ,затем водой аб-ре14 для удаления солей и остатков щелочи. После этого абгазы выводят на сжигание- они содержат метан, этан, пропан, олефинов практически нет. Реакционная масса выводится через боковой перелив вверху к-ны, охлаждается в хол-ке 15 и в С-16 отделяется от кат.комплекса. Состав рек-ной смеси (алкилат): бензол 45-55%, моноалкилбензол 35-40%. Диалкилбензол 8-12%; ПАБ,смолы, побочные продукты до 3%.
Алкилат идет на очистку от растворенной НCl и AlCl3.для этого его промывают водой а затем щелочью, а потом снова водой. Далее алкилат идет на ректификацию в систему ректиф. колонн. В первое к-не отгоняют бензол с водой; в след.к-не в вакууме отгоняют целевой продукт – моноалкилбензол. Далее в след. к-не отгоняют диалкилбензол и ПАБы. Остаток из последней кол-ны- это смолы, их сжигают. Выход целевого продукта достигает 94-95% при расходе AlCl3 от 5 до 10 кг на тонну моноалкилбензола
В-25. Производство изопарафинового алкилата и МТБЭ. Технологическая схема.
Алкилат- высокооктановый изокомпонент бензина. Производится С-алкилированием изобутана бутиленами и пропиленом. Алкилат- состоит практически нацело из изопарафинов, имеет высокое ОЧММ=90.Основным компонентом явл-ся изооктан (ОЧ=100). В общем виде: CnH2n+2 + CmH2m↔Cn+mH2(n+m)+2
Механизм карбоний ионный: CH3CH=CHCH3 + H+→ CH3CH2C*HCH3
CH3CH2C*HCH3 + (CH3)3CH → CH3CH2CH2CH3 + (CH3)3C+
RH + CH2=CHR*↔RR*CHCH3
Катализаторы: AlCl3, HF, H2SO4.
Алкилируются только изопарафины. В присутствии кат-ра карбкатион изомеризуется с перемещением заряда или метильных групп. Побочные реакции: получение высших УВ, деструкция карб-катиона, полимеризация олефина. Их подавляют избытком изопарафинов. Оптимальное соотношение изопарафина к олефину от 4:1 до 6:1. Т=0-100, Р=0,35-0,42 МПа. Сырье - бутан-бутеновая фракция крекинг газов. Реакционная масса – двухфазная система, которую эмульгируют при помощи мешалок. Большое значение имеет концентрация H2SO4 (лучшей явл-ся 98-100 %). Объемное отношение H2SO4 и УВ-дов = 1:1.
Реакционные узлы: двух видов, отличаются способом отвода выделяющегося тепла- охлаждением хладоагентом (аммиаком или пропаном) через теплообменную поверхность и охлаждением за счет испарения избыточного изобутана. В первом случае валкилаторе – контакторе вертикального или горизонтального типа, снабженном мощной мешалкой, имеются охлаждающие трубы, в к-рых хладоагент испаряется, пары которого направляются затем в холодильную установку, где они снова превращаются в жидкость. Реакторы второго типа – горизонтальные каскадные, в к-рых охлаждение реакционной смеси осуществляется за счет частичного испарения изобутана, что облегчает регулирование Т. Реактор имеет несколько зон смешения, разделенных перегородками, и двухсекционную зону отстоя. Бутилен подводится отдельно в каждую секцию, вследствие чего концентрация олефинов очень мала, это позволяет подавить побочные реакции. Применение каскадных реакторов упрощает и удешевляет установки.
Тех.схема установки производства алкилата. Т=5-15; Р=0,6-1 МПа. Исходная УВ-ная смесь поступает пятью параллельными потоками в смесительные секции реактора-алкилатора Р, в первую секцию вводятся H2SO4 и жидкий изобутан. Из отстойной секции Р выводятся продукты алкилирования, к-рые после нейтрализации щелочью и промывки водой идут в К-2 для отделения циркулируемого изобутана. Испарившиеся в реакторе изобутан и пропан через сепаратор Р-рессивер компрессором через холодильник подаются в колонну-депропанизатор К-1. Нижний продукт К-1-изобутан через кипятильник и теплообменник присоединяется к циркулирующему потоку изобутана из К-2. Нижний продукт К-2 поступает вколонну дебутанизатор К-3, а остаток К-3 – в К-4 для перегонки суммарного алкилата. С верха этой колонны отбирается целевой продукт – легкий алкилат, а с низа – тяжелый алкилат (компонент ДТ).
МТБЭ. МТБЭ явл-ся высокооктановым кислородсодер-жащим компонентом автобензина. По сравнению с другими компонентами МТБЭ обладает более высоким ОЧ и низкой температурой кипения (+ 55,2 С), что в совокупности позволяет повысить ОЧ преимущественно головных фракций базового бензина, тем самым и равномерность распределения детонационной стойкости по его фракциям. В товарные автобензины МТБЭ добавляют в количестве 5-15 %. Эфирсодержа-щие бензины характеризуются большой полнотой сгорания и меньшей токсичность выхлопных газов.
Процесс получения МТБЭ основан на реакции взаимодействия метанола с изобутиленом на ионообменном катализаторе кислотного типа : СН2 = С(СН3)(СН3) + СН3ОН ↔ СН3—С(СН3)(СН3)—ОСН3
Реакция синтеза МТБЭ из изобутилена и метанола протекает по цепному карбоний ионному механизму с выделением 66 кДж/моль тепла ,а её равновесие смещается вправо при повышении давления и снижения Т.
1. Первой стадией О- алкилирования метанола изобутиленом является протониро-вание последнего гидрид ионом кислотного катализатора : СН3—С(СН3)=СН2 + Н А ↔ СН3—С+(СН3)—СН3 + А+
2. Образовавшийся третичный бутеновый карбониевый ион вступает в реакцию с метанолом (при его избытке):
СН3—С(СН3)(СН3) —СН3 + СН3 ОН ↔ СН3–С(СН3)(СН3) –ОН–СН3 ↔ СН3–(СН3)(СН3)С–О–СН3 + Н-
3. Образовавшийся протон далее реагирует с изобутеном, как и в стадии 1.
4. Причиной обрыва цепи может стать возврат протона к катализатору Н- + А+ ↔ НА
Помимо основной целевой реакции О-алкилирования, при синтезе МТБЭ протекают побочные реакции:
- димеризация изобутена с образованием диизобутилена. Эта реакция идет с выделением большого количества тепла.
- гидратация изобутилена водой, содержащейся в исходном сырье с образованием изобутилового спирта;
- гидроконденсация метанола с образованием диметилового эфира.
Обычно реакцию образования МТБЭ ведут с небольшим избытком метанола чтобы ограничить побочные реакции. Получение МТБЭ- каталитический процесс. Применяют гомогенные (серная, фосфорная, соляная, борная кислоты) и гетерогенные катализаторы (цеолиты, оксиды алюминия, железа, никеля ,ионообменные смолы) кислотно-основного типа. Все существующие технологии синтеза МТБЭ обычно включают в себя три основных узла: реакторный узел, фракционирование и очистку отходящей фракции. Качество товарного МТБЭ колеблется в пределах 97 – 99% масс. В качестве промышленных реакторов используются трубчатые изотермические реакторы со съемом тепла циркулирующей водой, каскад адиабатических реакторов с промежуточным отводом тепла реакции или сочетание разных типов реакторных систем, реакторы реакционно-ректификационного типа, в которых отвод тепла осуществляется испарением потока флегмы. Сырьё: ББф кат.крекинга и пиролиза, и метанол.
Тех.схема. Исходная бутан-бутиленовая фракция через емкость Е поступает в верхнюю часть реактора форконтактной очистки. Очищенная смесь после нагрева в теплообменнике до 60ºС поступает в зону синтеза под каждый слой кат-ра реактора Р-1(2). В верхнюю часть реакционной зоны во избежание перегрева кат-ра подается свежий метанол с Т=50-60 С. Жидкие продукты, состоящие из МТБЭ с примесью метанола и УВ, выводятся из куба реактора и идут в отпарную колонну К-2 с паровым кипятильником на отпарку примесей. МТБЭ с куба К-2 после теплообменников и холодильников идет в товарный парк. Паровая фаза, состоящая из отработанной ББФ, метанола и следов МТБЭ, поступает на конденсацию МТБЭ в К-1. Конденсированный МТБЭ возвращается на верхнюю тарелку Р-1(2) в качестве холодного орошения. С верха К-1 отводятся пары ББФ и метанола, к-рые после конденсации идут в С-1, далее в К-3 разделяются экстракцией водой. Отработанная ББФ с верха К-3 после охлаждения в холодильниках идет в товарный парк. Отгонка циркуляциооного метанола от воды проводится в ректификационной колонне К-4, охлаждается и собирается в рефлюксной емкости С-3. Часть метанола исп-ся в качестве холодного орошения К-4, а остальная часть идет в Е. Вода из куба К-4 после охлаждения подается в экстрактор К-3 для отмывки метанола от ББФ.
В-26. Синтезы на основе α-оксидов. Продукты , технология, реакционные узлы.
Реакции идут с разрушением цикла. Этилен оксид химически активен, взаимодействует с веществами, содержащими подвижный атом водорода.
СН2СН2О + HA↔ HOCH2CH2A
Примеры синтезов:
1. Взаимодействие с водой СН2СН2О + НОН→ НОСН2СН2ОН
2. Вз-вие со спиртами и фенолами СН2СН2О + RОН→ НОСН2СН2ОR
3. Вз-вие с H2S и RSH: СН2СН2О+ H2S → НОСН2СН2SH
СН2СН2О+ RSH → НОСН2СН2SR
4. Вз-ие с карбоновыми и неорганическими кислотами: СН2СН2О + RCOOH→ HOCH2CH2OCOR
5. Вз-ие с аммиаком, аминами и амидами: СН2СН2О + =NH→ HOCH2CH2N=
6. Реакции с расширением цикла: с СО2 и СН3СООН.
Механизм: эти реакции часто идут без кат-ра, но сильно ускоряются кислотами или щелочами. При взаимодействии с О-сод. соед-ми, H2S, RSH или амидами наиболее типичными кат-ми явл-ся основания (NaOH, Na2CO3, NR3)- катализ основный или нуклеофильный. NaOH + НА → Н2О + Na+ + А-
![]()
По такому же механизму протекает и не каталитическая реакция. Нуклеофилом явл-ся сама молекула реагента, но реакция идет медленнее. С достаточной скоростью она протекает при Т=180-220ºС, при нуклеофильной реакции 100-150ºС. Кислотный катализ тех же реакций протонными кислотами эффективен в сильнополярных средах (Н2О, низшие спирты).
![]()
В малополярной среде протонные кислоты присоединяются к α-оксидам и становятся неактивными – применяют гетерогенные кат-ры кислотного типа (BF3, SnCl4, Al2O3, HF). Реакции кислотного катализа протекают при Т от 20-40 до 100-150.
Продукты:
1) Гликоли- прим-ся для производства антифризов, взрывчатых веществ, растворителей, полимерных материалов.
2) Этаноламины- прим-ся для произ-ва взрывчатых веществ и для очистки газов от кислотных примесей.
3) Неионогенные ПАВы- применяют как компоненты моющих средств.
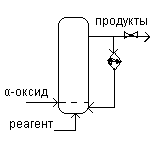
Технология и реакционные узлы. Все производства на основе α-оксидов по технологическим признакам делятся на три типа :
1) Реакции осуществляющиеся при большом избытке реагента – произ-во гликолей и эфиров. Теплота реакции воспринимается избыточным реагентом. Процесс осущ-ся в адиабатических и полностью гомогенных условиях.
(А) (Б) (В)
![]()
 (Г)
(Г)
Рис.А: Непрерывно действующая реакционная колонна, не имеющая поверхности теплообмена. Исходная смесь, предварительно подогретая паром, подается с верху и поступает в низ колонны по центральной трубе, в к-ой она еще подогревается реакционной массой. «-»: снижение селективности за счет наличия продольного перемешивания продукта.
2)Для непрерывного осуществления таких реакций в более интенсивном режиме, больше подходят кожухотрубчатые реакторы (Рис.Б). Применяется для реакций, в к-рых мольное отношение α-оксида к реагенту поддерживают от 1-(4-5) до (2-3)-1: синтез этаноламинов, тиогликолей, тиоэфиров. Применяется реакционный узел с циркуляцией реакционной массы через выносной холодильник (Рис.В). «-»: аппарат применим только для тех реакций, в к-рых последовательное превращение не играет существенной роли: получение этиленкарбоната, тиоспиртов и тиоэфиров. 3) Реакции, в к-рых мольное отношение этиленоксида к реагенту превышает 3:1 – синтез полигликолей и неионогенных ПАВ. Процесс осуществляется периодическим способом в аппарате с разбрызгиванием жидкой реакционной массы в атмосферу газообразного этиленоксида (Рис.Г). Охлажденную в кубе жидкость непрерывно прокачивают через спец.форсунки и впрыскивают в газообразное пространство реактора, куда вводят этиленоксид. Образуются капельки ПАВ, к-ые оседают в жидкую фазу.
В-27.Процессы галогенирования. Классификация реакций их галогенирующая характеристика.
Галогенирование – процессы в результате которых в органические соединения вводятся атомы галогена.
В зависимости от типа галогена различают реакции фторирования, хлорирования, бромирования и йодирования.
Это один из самых распространенных процессов и этим путем получают различные виды продуктов:
1)Хлорорганические промежуточные продукты – введение в молекулу достаточно подвижного атома хлора позволяет при дальнейших превращениях получить ряд ценных веществ (1,2-дихлорэтан, хлоргидрины, алкилхлориды).
2)Хлор и фтор органические мономеры (винилхлорид, винилиденхлорид , тетрафторэтилен)
3)Хлорорганические растворители (дихлорметан, тетрахлорметан, три- и тетрахлорэтилены)
4)Хлор и бром органические пестициды (гексахлорциклогексан, хлорпроизводные кислот и фенолов)
Кроме того хлорпроизводные используются как холодильные агенты, а также в качестве пластификаторов и смазочных масел.
Характеристика процессов галогенирования.
Заместительное галогенирование: Состоит в замещении на атомы галогена других атомов или групп. Наибольшее значение имеет замещение атомов водорода , которое может происходить как по насыщенным, так и ненасыщенным атомам углерода или в ароматическое ядре. RH + Cl2 → RCl + HCl
Замещение одного атома галогена на другой имеет значение для получения фтор, бром, и йод производных. Из наиболее доступных хлорорганических соединений.
CCl4 + 2HF → CCl2F2 + 2HCl ; RCl + NaBr → RBr + NaCl
ROH + HCl → RCl + H2O ;
RCOOH + COCl2 → RCOCl + CO2 + HCl
Присоединительное галогенирование: Присоединение галогенирующих агентов по ненасыщенным связям .
СН2=СН2 + Cl2 → ClСН2 - СН2Cl ;
СН≡СН + 2 Cl2 → СНCl2 - СНCl2
Галогенводороды присоединяются по двойной и тройной связям (гидрогалогенирование), а олефины также вступают в реакцию хлоргидринирования.
СН2=СН2 +HCl→СН3СН2Cl; СН≡СН+ HCl →СН2=СНCl
СН2=СН2 + Cl2 + Н2О → СН2Cl - СН2ОН + HCl
Особый случай аддитивного хлорирования- присоединение Cl атомам, находящимся в низшем валентном состоянии: СО + Cl2 = COCl2 – фосген.
Реакции расщепления хлорпроизводных. Наиболее легко происходит дегидрохлорирование. Из-за предпочтительности протекания этой реакции другие процессы расщепления наблюдаются только при высокой температуре у перхлорпроизводных (дехлорирование, расщепление по С – С связям которое происходит под действием хлора ) (хлоролиз) или при повышенной температуре (пиролиз).
Дегидрохлорирование:ClСН2 - СН2Cl → СН2=СН2 + HCl
Дехлорирование: СCl3 - СCl3 → СCl2 = СCl2 + Cl2
Хлоролиз: СCl3 - СCl3 + Cl2 → 2СCl4
Пиролиз: СCl3 - СCl2 - СCl3 → СCl4 + СCl2= СCl2
Термодинамика галогенирования.
Тепловой эффект уменьшается в ряду F> Cl2>Br2>I.
При фторировании и йодировании выделяется тепло, превышающее энергию разрыва связи С – С и С – Н. Это приведет к глубокому разложению органического вещества. В следствии этого технология фторирования значительно отличается от хлорирования.Йодирование напротив протекает с очень небольшим выделением тепла и является обратимым процессом
Все реакции экзотермичны, здесь тепловой эффект значительно ниже чем для свободных галогенов.
Галогенирующие агенты.
Наибольшее значение получили свободные галогены и безводные галогеноводороды:
F Tк= -188 0C НF Tк = 19,40 C
Cl Tк = -35.5 0C HCl Tк = -83,7 0C
Br Tк = 58.8 0C НBr Tк = - 67 0C
Все они растворимы в органических жидкостях. Их растворимость уменьшается в ряду
Br2> Cl2>F2 ; НBr> HCl> НF Эти свойства важны для проведения жидкофазных процессов галогенирования. Все галогены имеют резкий запах, раздражают слизистые оболочки глаз и дыхательных путей. Свободные галогены кроме того обладают удушающим действием, особо опасны F и НF они способны разъедать кожные покровы и костную ткань.
Все галогенирующие агенты агрессивны к материалу аппаратуры, причем их корродирующее действие возрастает в присутствии влаги, поэтому в процессах фторирования для производства аппаратуры применяют медь, никель, а при хлорировании и бромировании защищают стальной корпус эмалями, свинцом, керамикой. Используют также специальные сорта сталей, графит, стекло, а для изготовления труб – свинец. Для снижения коррозии галогенирующие агенты и органические реагенты подвергают сушке.
В-28. Получение хлористого винила оксихлорированием.
В процессах заместительного хлорирования и расщепления хлорпроизводных всегда образуется HCl, которую утилизируют в виде соляной кислоты.Устронение побочного образования соляной кислоты решается за счет окислительного хлорирования. В основе этого процесса лежит реакция Дикона: 2HCl + 0,5О2 ↔ Н2О + Cl2
В следствии экзотермичности процесса равновесие сдвигается вправо при понижении т, но известные кат-ры на основе СuCl2 позволяют работать только при Т=200 – 4000C. В этих же условиях протекают реакции хлорирования, а при совмещении обоих процессов в одном аппарате окислительное хлорирование при помощи HCl и О2 становится не обратимым.
RH + HCl + 0.5O2 → RCl + H2О
Кроме основной реакции происходит побочное окисление углерода кислородом, гидролиз хлорпроизводных и дегидрохлорирование, поэтому этот процесс можно использовать для стабильных исходных веществ (метан, этилен, бензол) впервые был применен для получения фенола .При окислительном хлорировании этилена (t = 210 – 2800C) происходит присоединение Cl по второй связи, а не его замещение. Хлорирование осуществляют не хлором, а непосредственно СuCl2, который регенерируется под действием HCl и О2.
Технология сбалансированного по хлору синтеза винилхлорида.
Этот процесс является комбинацией трех реакций:
1)Прямое аддитивное хлорирование этилена в 1,2 – дихлорэтан.
СН2=СН2 + Cl2 →ClСН2 - СН2Cl
2)Термическое дегидрохлорирование 1,2 – дихлорэтана с образованием винилхлорида.
2ClСН2 - СН2Cl →2СН2=СНCl + 2НCl
3)Окислительное хлорирование этилена в 1,2 – дихлорэтан.
СН2=СН2 + 2НCl +0,5 O2 → ClСН2 - СН2Cl +Н2О
В результате из этилена, хлора и кислорода получается винилхлорид, причем хлор полностью расходуется и не образуется НCl. Технологическая схема производства винилхлорида.
Прямое хлорирование этилена до 1,2 – дихлорэтана проводят в колонном хлораторе 1 куда этилен и хлор подают через соответствующие барбатёры. В колонне сохраняется постоянный уровень жидкости, в которой растворен катализатор FeCl3. тепло реакции отводится за счет испарения 1,2 – дихлорэтана при этом его пары конденсируются в конденсаторе 2. конденсат попадает в сборник 3, откуда часть его возвращается в колонну, а остальное идет на ректификацию. В сборнике 3 от конденсата отделяют остаточные газы, которые во избежание потерь 1,2 – дихлорэтана дополнительно охлаждают рассолом в холодильнике 2. затем направляют на очистку и выводят в атмосферу.
Стадия оксихлорирования проводится в реакторе 5 с псевдоожиженным слоем катализатора. Давление в реакторе = 0,5 МПа, температура = 250 0С. Этилен, рециркулирующий газ и НCl предварительно смешиваются в трубе, затем в смесителе 4 добавляют технический кислород. Способ смешивания и состав смеси подбирают таким образом, чтобы обеспечить взрывобезопасные условия. В реакторе 5 тепло отводится за счет испарения водного конденсата. В результате получается технический пар, который используют для собственных нужд. Реакционные газы, состоящие из не превращенного этилена, кислорода и НCl, а также паров 1,2 – дихлорэтана охлаждают в холодильнике 6 смесью воды и 1,2 – дихлорэтана, которая циркулирует через холодильник 7 .
Частично охлажденная смесь очищается от НCl и СO2 в горячем щелочном скруббере и окончательно охлаждают в холодильнике 10. Конденсат отделяют от газов в сепараторе 11. После чего в компрессором 13 возвращают на оксихлорирование.
Часть газа во избежание накопления инертных примесей направляют в общую линию отходящих газов и после очистки выводят в атмосферу.
Конденсат из сепаратора 11 идет в сепаратор 12 где 1,2 – дихлорэтан отделяется от воды. Воду используют для разбавления щелочи, которой очищают газ в скруббере 9. 1,2 – дихлорэтан полученный при оксихлорировании содержит воду, поэтому его осушают в колонне азеотропной осушки 14. После чего оба потока 1,2 – дихлорэтана с прямого хлорирования и оксихлорирования объединяют и в ректификационной колонне 16 1,2 – дихлорэтан отгоняют от высших хлоридов.
Пиролиз 1,2 – дихлорэтана проводят в трубчатой печи 19 при давлении = 1,5 – 2 МПа и температуре = 500 0С, при этом получается винилхлорид. Реакционные газы охлаждаются в холодильнике 20 циркулирующим 1,2 – дихлорэтаном, а затем в конденсаторе-холодильнике водой. Затем смесь поступает в ректификационную колонну 21 . где винилхлорид очищается от HCl. HCl возвращается на оксихлорирование. Кубовая жидкость колонны 21, состоящая из винилхлорида и не превращенного 1,2 – дихлорэтана, направляется в колонну 22 где от винилхлорида отделяют 1,2 – дихлорэтан. Полученный винилхлорид содержит 99,9 % целевого продукта.
В-29. Процессы фторирования.
Из реакций фторирования главное промышленное значение имеют следующие:
1)Действие молекулярного фтора и некоторых фторидов металлов, находящихся в высшем валентном состоянии (СоF3 , AgF2), при этом происходит замещение на фтор атомов водорода.
2)Действие HF и его солей, в которых атомы металла находятся в нормальном состоянии (AgF, HgF2, SbF3 ), при этом на фтор замещаются в основном атомы хлора.
Фторирование фтором и высшими фторидами металлов. Прямое действие фтора на органические вещества приводит к бурной реакции, которая сопровождается вспышками и взрывами. В результате получаются НF и продукты разложения органических молекул (С , СF4). Такое направление реакции обусловлено высокой экзотермичностью, которая превосходит энергию разрыва связей С – С и С – Н. Более спокойно реакция идет при разбавлении фтора. Обычно разбавляют азотом. Механизм фторирования сильно отличается от хлорирования из – за очень слабой электрофильности молекулы фтора. Поэтому протекают только радикально – цепные реакции. Инициаторов не требуется, и цепь зарождается самопроизвольно.
RH + F2 →R* + Н*F2 → R* + HF + F*
RH + F* → R* + HF
R* + F2 → RF + F*
Существует 3 метода фторирования:
Каталитическое фторирование. Осуществляется в аппарате с медной стружкой, покрытой слоем серебра. В аппарат подают фтор и УВ, разбавленные азотом. Роль меди состоит в аккумулировании тепла, а серебро переходит под действием фтора в AgF2 – фторид серебра, который легко фторирует углеводород. Однако побочно происходит прямое взаимодействие углеводорода со фтором, вследствии чего выход целевого продукта очень мал (40 – 80 %).
Металлфторидный процесс. Наиболее распространен, основан на применении высших фторидов металлов (СоF3, MnF3), которые взаимодействуют с УВ мягко и с небольшим выделением тепла. Образующуюся соль вновь регенерируют под действием фтора.
RH + 2 CoF3 → RF + HF + 2CoF2
2CoF2 + F2 → 2CoF3
УВ принимает участие только в одной реакции, меньший тепловой эффект, вследствие чего увеличивается выход продукта.
Реактор представляет собой горизонтальную стальную трубу с лопастной мешалкой. Труба заполнена наполовину фторидом кобальта. В левую часть трубы поступают пары органического реагента с 5-10-кратным избытком азота. С другого конца реактора отводят образующиеся продукты, которые сначала попадают в выводную трубу, где освобождаются от частиц фторида кобальта, а затем идут на охлаждение и разделение. Реакцию ведут пока 50% CoF3 не превратится в CoF2. Т в реакторе поддерживают не одинаковую: на входе – 150 – 2000С, на выходе – 300 – 3800С т.к. УВ фторируется в начале очень быстро и для этого нужна низкая Т. Электрохимическое фторирование-
Сущность метода: при электролизе безводного HF – фторида водорода, выделяющийся на аноде фтор реагирует с УВ, который растворен в жидкости. Благодаря протеканию реакции в жидкой фазе и при интенсивном перемешивании достигается хороший теплоотвод и появляется возможность в регулировании процесса. При этом не приходится предварительно получать и очищать молекулярный фтор. Метод в основном применяется при синтезе карбоновых кислот, простых и сложных эфиров, аминов, сульфидов и других соединений, растворимых в жидком фториде водорода.
Получаемые продукты. Фторорганические мономеры(тетрафторэтилен и монохлортрифторэтилен. винилхлорид С2H3F, винилиденфторид C2H2F2.
Фреоны (хладоны)- фторхлорпроизводные метана и этана. Это газообразные вещества или низкокипящие жидкости, обладают слабым запахом, очень мало токсичны, совершенно не горючие. Применяются: в холодильных машинах, в аэрозольных распылителях.
Важнейшие фреоны в промышленности получают замещением атомов хлора на фтор в следующих продуктах: хлороформ, тетрахлорметан, тетра-, пента- и гексахлорэтан.
Примеры фреонов:
СНCl3 → CHCl2F →CHClF2 →CHF3
21 (8.9) 22(-40) 23(-82.2)
CCl4 → CCl3F → CCl2F2 → CClF3
11 (23.7) 12(29.8) 13 (-81.5)
Наибольшее значение в технике имеют фреоны 12, 22, 113. При глубоком охлаждении используют фреоны 13 и 23.
Фреон 12 (дифтордихлорметан)- образуется из тетрахлорметана и безводного фторида водорода в присутствии галогенидов сурьмы. Реакцию ведут при 1000С Р = 3МПа. В этих условиях все вещества участвующие в реакции находятся в жидком состоянии.
Технологическая схема производства фреона 12.
Тетрахлорметан и жидкий фторид водорода подаются насосами 1 и 2 в реактор 3, где находится жидкий катализатор: смесь хлоридов и фторидов трех- и пятивалентной сурьмы. Также в реактор небольшими порциями подается хлор. Реактор представляет собой стальной аппарат с антикоррозийным покрытием. Снабжен паровой рубашкой, дефлегмирующей колонкой 4 и обратным конденсатором 5. Образующийся НCl уносит с собой пары органических реагентов и фторид водорода. В колонке 4 происходит конденсация паров. При этом тетрахлорметан и монофтортрихлорметан возвращается в реактор. Для создания флегмы конденсируют часть паров CF2Cl2 в конденсаторе 5 и возвращают на орошение в колонку 4. Парогазовая смесь, выходящая из конденсатора 5 содержит главным образом НCl и СF2Cl2 с примесью CFCl3 и HF. После снижения давления до атмосферного HF отделяется в башне 7, которая заполнена кусками фторида калия. KF взаимодействует с HF образуя KHF2, который можно использовать для получения фтора методом электролиза. Дальнейшую очистку от HF осуществляют с применением концентрированной соляной кислоты. Очистка идет путем абсорбции в скруббере 8, который орошается водой и в скруббере 9, который орошается разбавленной щелочью. Осушку газа проводят с помощью концентрированной серной кислоты циркулирующей в колонне 10. Для разделения фторхлорпроизводных используют низкотемпературную ректификацию. Пары сжимают компрессором 13 до Р = 1-1,2 МПа и охлаждают в холодильнике11 до Т = (-10) – (-15)С. Образующийся конденсат поступает на низкотемпературную ректификацию. Легкая фракция состоит из СClF3 – побочный продукт производства. Тяжелый остаток содержит СFCl3, который возвращают в реактор. Целевая фракция фреона 12 получается в жидком виде под давлением. Для ее использования в качестве хладагента ее осушают путем вымораживания влаги и обработкой цеолитами.
В-30.Процессы дегидрирования и гидрирования.
Дегидрирование – это химический процесс связанный с отщеплением атома водорода от органического соединения.
Классификация реакций дегидрирования.
1. С-С дегидрирование парафинов. Образуются соединения с двойной С=С связью и в дальнейшем- диены.
СН3 – СН2 – СН2 – СН3 → СН3 – СН2 – СН = СН2+Н2 →СН2 = СН – СН = СН2 +Н2
Боковые цепи аренов также могут дегидрироваться с образованием веществ типа стирола.
2. Дегидрирование по С-О связи - характерно для первичных и вторичных спиртов, из которых образуются альдегиды и кетоны.
R–CH2OH →RCOH + Н2 ; R2CHOH → R2CO + Н2
3.Дегидрирование по С – N связи. Характерными примерами явл-ся отщепление Н2 от первичных аминов с образованием нитрилов.
4.Дегидроциклизация, когда из парафинов путем замыкания цикла получаются арены.С6Н10 → С6Н6+ 2Н2
Это важнейший путь ароматизации узких фракций нефти.
5.Дегидроконденсация приводит к образованию ди- и полиядерных соединений. 2С6Н6 → дифенил
Классификация реакции гидрирования.
1.Присоединение Н2 по ненасыщенным связям. Могут происходить с присоединением водорода по тройной связи , по двойной Сар = Сар ядре, по С=О, С≡N.
RCH = CH2 +H2 ↔ RCH2 – CH3
RR’CO +H2 ↔ RR’CHOH ; RCN + 2Н2 ↔ RCH2NH2
2.Гидрирование, сопровождающееся отщеплением воды или других веществ, не содержащих углерода. Это реакции восстановления органических соединений (гидрирование карбоновых кислот в спирты; спиртов а УВ; амидов кислот и нитросоединений в амины).
RCOOH + 2H2 → RCH2OH + H2O
ROH + H2→RH +H2O ;
RCONH2+2H2→RCH2NH2+H2O;RNO2+3H2→RNH2+H2O
Могут отщепляться не только вода, но и HCl, NH3, H2S:
RCOCl + H2 →RCHO + HCl ; RSH + H2 → RH + HS2
3.Гидрирование, сопровождающиеся расщеплением УВ. К ним способны УВ с открытой цепью: нафтены или арены с боковой цепью: RCH2R’ + H2→ RCH3 + R’H
Эти реакции обратны процессам дегидроконденсации и дегидроциклизации. Особым типом гидрирования и дегидрирования является перераспределение атомов водорода между молекулами. Одна из них отщепляет водород, а другая присоединяет его.
Катализаторы. 1.Ме 4 группы (Fe, Co,Pd,Pt,Ni);Ag и Ni, сплавы этих Ме.
2.Оксиды Ме (MgO; Zno;Cr2O3; Fe2O3).
3. Сложные оксидные и сульфидные кат-ры. Они состоят из смеси оксидов и сульфидов Ме (медьхромоксидные (CuO·Cr2O3), цинкхромоксидные (ZnO·Cr2O3), кобальтмолибденооксидные (СоО·МоО3). Эти вещества часто наносят на пористые носители, затем к ним добавляют различные промоторы.
Механизм реакций гидрирования
К-СН2-СН2- + Н2···К ↔ К-СН2-СН3 + КН ↔ СН3-СН3 + 2К
дегедрирования
RСН2-СН3-2К ↔ RCKH-CH3 + K↔ RCKH-CH2↔RCH=CH2
Кинетика реакций гидрирования и дегидрирования. Скорость этих реакций зависит от диффузионных и кинетических факторов. Диффузионные факторы играют меньшую роль, если осуществляется интенсивное перемешивание и процесс протекает при пониженной Т. Энергия активации дегидрирования довольно высока (от 100 до200 кДж/моль). Это определяет сильную зависимость скорости дегидрирования от Т. Способность к дегидрированию на оксидных кат-рах уменьшается в ряду: спирты > амины > алкилбензолы > парафины.
Скорость дегидрирования увеличивается при удлинении и разветвлении УВ-ной цепи.
Процессы гидрирования обычно осуществляют в условиях когда равновесие смещено вправо и можно пренебречь обратной реакцией дегидрирования. С такими кат-рами как Pt, Pd, Ni скорость реакции при умеренных Т (100ºС) не зависит от Рпарц водорода. На оксидных же кат-рах сорбция водорода менее значительна чем на Ме, поэтому зависимость скорости реакции от Рпарц водорода линейна, поэтому процесс ведут при высоком Р и избытке водорода. Т на скорость гидрирования оказывает небольшое влияние и обычно при повышении Т процесса на 30 – 50ºС скорость примерно удваивается. По способности к гидрированию УВ располагаются в ряду: олефины > ацетилен и его производные > арены и альдегиды > кетоны > нитрилы > карбоновые кислоты. В-
В-31. Дегидрирование олефинов.
В термодинамическом отношении столь же неблагоприятно, как и дегидрирование алкилароматических у/в. Поэтому и здесь для повышения равновесной ст.конв. при допустимой темп-ре (≈600ºС) приходится разбавлять реагирующую смесь водяным паром.
н- Бутены или изопентены, поступающие на дегидрирование, независимо от их происхождения (из прод-в пиролиза, крекинга или дегидрирования соответствующих парафинов) представляют собой смесь изомеров, причём дегидрируются только α-олефины:
-Н2
СН2=СН-СН2-СН3 ↔ СН2=СН-СН=СН2
Для β-изомеров необходима предварительная изомеризация с перемещением двойной связи или образованием поверхностного радикала с делокализованными электронами:
СН3-СН=СН-СН3 ↔ СН2=СН-СН-СН3 -Н•
-Н2
СН3-СН=СН-СН3 ↔ [СН3-СН-СН-СН2]• → СН2=СН-СН=СН2
Кроме этих целевых реакций при дегидрировании протекают побочные процессы крекинга, скелетной изомеризации и коксообразования. В рез-те крекинга из олефинов получаются метан и у/в С2 и С3. Изомеризация н-бутена ведёт к образованию изобутена, но эта р-я особенно нежелательна для изопентенов, когда получившиеся н-пентены могут дальше дегидрироваться в пентадиен-1,3 (пиперилен), а последний способен замыкать цикл с образованием циклопентадиена:
(СН3)2С=СН-СН3
↔
СН3-СН2-СН=СН-СН3
↔
СН2=СН-СН=СН-СН3
→
Отрицательная роль последних р-ий состоит не столько в снижении
селективности, сколько в получении примесей, затрудняющих очистку и выделение целевых веществ.
При дегидрировании олефинов образуются продукты уплотнения и кокс. Считается, что главным источником являются диены, кот. склонны к реакциям конденсации с образованием циклических сис-м (диеновый синтез с последующей дегидроконденсацией ароматических соединений).
Наконец, при дегидрировании олефинов за счет образующегося Н2 получается небольшое кол-во парафинов, которые крекируются легче, чем соответствующие олефины. Часть у/в и кокса подвергается также конверсии водяным паром, вследствие чего в газе содержатся оксиды углерода.
Т.о., схема превращений при дегидрировании олефинов такова:

Итак, имеются паралл. и последоват. пути обр-я побочных в-в, причём как и при дегидрировании алкилароматических у/в, селективность растет при снижении 2-х факторов- темп-ры (из за более высокой энергии активации побочных реакций) и ст.конв.; выбор этих вел-н обусловлен экономическими соображениями.
В соответствии с изложенным, kat дегидрирования олефинов должны ускорять преимущественно дегидрирование и изомеризацию с перемещением двойной связи, но должны быть мало активными в отношении крекинга, скелетной изомеризации и коксообразования. В наст. время лучшими явл-ся кальцийникельфосфатные kat, имеющие в СССР наименование ИМ-2204. Их состав соответствует формуле Са8Ni(PO4)6, они содержат промотирующую добавку (≈2% Cr2O3), выпускаются в формированном виде для работы в стационарном слое.
Характерная особенность этих контактов- быстрое закоксовывание и потеря активности, вследствие чего требуется часто выжигать кокс. Периоды дегидрирования и регенерации kat чередуют каждые 15 мин, предварительно продувая реактор водяным паром.
Дегидрирование ведут. разбавляя исх. смесь водяным паром в объёмном соотношении 20:1, при объёмной ск-ти по газообразному у/в 150-200ч-1 и общем давлении, только немного превышающем атмосферное (чтобы преодолеть гидравлическое сопротивление слоя kat и последующей аппаратуры). Оптимальная темп-ра при дегидрировании н-бутенов 600-650ºС; ст. конв. при этом 40-50%, а селективность по бутадиену-1,3 85%. Для изопентена, более реакционно-способного и более склонного к побочным р-ям, темп-ра снижается до 550-650ºС; тогда ст. конв. достигает 40%, а селективность 82-84%.
В-32.Дегидрирование парафинов.
Дегидрированием парафинов получают изобутен и высшие олефины. При производстве бутадиена-1,3 и изопрена эта реакция является 1-й стадией процесса. Допустимая температура 600ºС процесс ведут без разбавителей при давлении чуть выше атмосферного.
Катализаторы дегидрирования должны быть активными по отношению к основной реакции и по возможности не ускорять процессы крекинга, изомеризации и закоксовывания. Лучшими кат-рами являются оксидные алюмохромовые катализаторы на основе Al2O3. Такие кат-ры содержат 10-40% Сr2О3, 2-10% оксидов щелочных металлов Na2O, К2О, ВеО. Оксиды металлов служат для нейтрализации кислотных центров Al2O3, вызывающих крекинг и изомеризацию. Эти кат-ры чувствительны к влаге, поэтому исходные фракции С4 и С5 осушают до содержания влаги не более 1мг/м3. Ввиду высокой эндотермичности процесса и отсутствии разбавителя теплоносителя широко используют системы с псевдоожиженным слоем катализатора. В них применяется регенеративный принцип использования тепла и непрерывная регенерация кат-ра. В таких реакторах происходит значительное перемешивание смеси, а это снижает производительность и селективность, поэтому реактор и регенератор снабжают тарелками провального типа, что улучшает показатели процесса.
Технологический процесс состоит из 3-х стадий:
1. Дегидрирование парафина с регенерацией катализатора.
2. Выделение бутан-бутеновой или пентан-пентеновой фракции.
3. Разделение этой фракции с получением бутенов или изопентенов.
Дегидрирование высших парафинов. При этом процессе получают олефины С12-С18. Алюмохромовый кат-тор здесь не работает, поэтому применяют Рt с добавками металлов и щелочей, нанесенных на оксид алюминия, силикагель или цеолиты. Реакционную смесь разбавляют водородом, который предотвращает быстрое закоксовывание кат-ра, а также развитие побочных реакций. Мольное соотношение водорода к парафинам составляет (6-8):1, Р=0,2 - 0,4 МПа. Селективность в этих условиях 89-93%. Дегидрирование проводят в реакторе со стационарным слоем кат-ра при Т=460-500ºС.
После отделения водорода и продукта крекинга целевой продукт перерабатывают двумя методами: 1). Направляют на алкилирование бензола с последующим получением сульфанола. 2). Разделяют на нормальные парафины и нормальные олефины и затем используют олефины для других синтезов.
Одностадийное дегидрирование парафинов в диены.
C4H10 ↔ C4H8 + Н2 ↔ C4H6 + Н2
Равновесный состав этой системы зависит от Т и Р. При повышении Т равновесная концентрация бутана резко падает, содержание н-бутенов проходит через максимум, а кол-во бутадиена-1,3 растет, но незначительно ввиду одновременного образования водорода на обоих стадиях. Из этого следует, что для одностадийного процесса следует выбирать более высокую Т, чем на первой стадии дегидрирования парафинов и пониженное Рпарц реагентов. Также требуется кат-р, который ускорял бы обе реакции дегидрирования (алюмохромовый кат-р). Так как с этим кат-ром нельзя использовать водяной пар в качестве разбавителя, был разработан процесс, идущий при пониженном Р (0,015-0,02 МПа) и Т=580-600 º С. Из-за применения вакуума реакторы с движущимся слоем кат-ра оказались непригодными. Сильное отложение кокса и частая регенерация кат-ра обусловили использование регенеративной системы Гудри. Реакционный узел включает ряд блоков, состоящий из 5-8 горизонтальных реакторов со стационарным слоем кат-ра. Каждый реактор работает периодически по регенеративному принципу использования тепла, в период выжигания кокса кат-р разогревается до 600ºС, затем удаляют газы сгорания при помощи вакуума 1,5-2 минуты и проводят дегидрирование. В это время используется тепло насадки для проведения эндотермического процесса и когда тепло насадки опустится до минимальной (580ºС), реактор продувают перегретым водяным паром для вытеснения УВ (также 1,5-2 мин.) и вновь проводят регенерацию кат-ра. Чтобы кат-р не слишком быстро охлаждался, к нему добавляют гранулы прокаленного глинозема (насадка), играющего роль аккумулятора тепла, но и в этом случае стадии дегидрирования и регенерации длятся по 5-9 минут, с общей длительностью цикла работы реактора 15-20 минут. Все переключения потоков происходят автоматически и благодаря наличию в блоке 5-8 реакторов создается непрерывный поток исходных веществ и получаемых продуктов. Степень конверсии 20-30%, селективность 55%. Контактный газ содержит 11% масс. бутадиена-1,3 и 25-30% бутена. Из-за проведения обеих стадий дегидрирования не в оптимальных для них условиях, селективность здесь ниже, чем при двухстадийном процессе, но это компенсируется снижением капиталовложений и энергетических затрат благодаря сокращению некоторых стадий производства.
В-33.Гидрирование.
В ряде случаев гидрирование приводит к восстановлению кислородсодержащих веществ
Классификация реакции гидрирования.
1.Присоединение водорода по ненасыщенным связям. Могут происходить с присоединением водорода по тройной связи , по двойной Сар = Сар ядре, по С = О, С тройная связь N.
RCH = CH2 +H2 ↔RCH2 – CH3
ацетилен +H2 ↔CH2 = CH2
бензол +3Н2 ↔ циклогексан
RR’CO +H2 ↔RR’CHOH
RCN + 2Н2 ↔RCH2NH2
2.Гидрирование, сопровождающееся отщеплением воды или других веществ, не содержащих углерода. Это реакции восстановления органических соединений (гидрирование карбоновых кислот в спирты; спиртов а углеводороды; амидов кислот и нитросоединений в амины).
RCOOH + 2H2 →RCH2OH + H2O
ROH + H2 →RH +H2O
R – CONH2 +2H2→RCH2NH2 + H2O
RNO2 + 3H2 →RNH2 + H2O
Могут отщепляться не только вода, но и хлорид водорода, NH3, H2S:
RCOCl + H2 →RCHO + HCl
RSH + H2 →RH + HS2
3.Гидрирование, сопровождающиеся расщеплением углеводорода. К ним способны углеводороды с открытой цепью, например нафтены или арены с боковой цепью:
RCH2R’ + H2→RCH3 + R’H
циклогексан + Н2 → CH3-CH2-CH2-CH2-CH2-CH3
бензол –R +H2 → бензол + RH
Все реакции гидрирования являются экзотермичными. Тепловой эффект является наиболее высоким для соединений с тройной связью. Для ароматических соединений он меньше чем для олефинов, что обусловлено нарушением устойчивой ароматической системы. Гидрирование альдегидов более экзотермично чем гидрирование кетонов.
Реакционные узлы


а-колонна переодического действия с выносным охлождением для процесса с суспендированым катализатором, б- коскад колонн непрерывного действияс внутренним охлождением для процесса с суспендированым катализатором, в-колонна непрерывного действия с внутренним охлождением для процесса со стоционарным катализатором, г- колонна непрерывного действия с охлождением холодным водородом для процесса со стационарным катализатором.
Технологическая схема и основы производства высших жирных кислот.
Свежий и рециркулирующий водород сжимают компрессорами 1 и 2 до 30 МПа, смешивают и подогревают в теплообменнике 3 (горячими реакционными газами) и в трубчатой печи 4 (топочными газами). Эфир подают на реакцию насосом 5 через паровой подогреватель 6, а катализатор (в виде суспензии в высших спиртах) – насосом 16. Перед реактором 7 все компоненты смешивают; за счет тепла водорода смесь нагревается до необходимой температуры (300оС). Продукты реакции состоят из высшего спирта, метанола, небольших количеств непревращенного эфира и побочных продуктов – углеводородов и воды. Вместе с катализатором и избыточным водородом эта смесь выходит с верха реактора 7 и разделяется в горячем сепараторе 8 на жидкую и газовую фазы. Газовая фаза отдает свое тепло водороду в теплообменнике 3 и дополнительно охлаждается водой в холодильнике 9; образовавшийся конденсат отделяют от водорода в сепараторе 10 высокого давления. Этот водород возвращают на гидрировании. Жидкую фазу из сепаратора 8 дросселируют до давления, близкого к атмосферному, охлаждают водой в холодильнике 11 и отделяют от выделившегося от дросселированния газа в сепараторе 12 низкого давления. Жидкость поступает на центрифугу 13, где оседают более крупные частички катализатора, захватывающие с собой примерно трехкратное количество спиртов. Этот шлам шнеком 14 транспортируют в смеситель 15, куда добавляют свежий катализатор. Полученную смесь подают в реактор 7 насосом 16. Таким путем 85% катализатора циркулирует и возвращается в процесс. Остальное его количество находиться в чрезмерно измельченном виде (в результате истирания зерен) и выходит из центрифуги 13 вместе с главной массой продуктов, отфильтровываясь от них на фильтр-прессе 17. Этот катализаторный шлам выбрасывают. Жидкую фазу из сепаратора 10 дросселируют и отделяют от газа в сепараторе 18 низкого давления. Газ объединяют с газом из сепаратора 12 и используют в качестве топливного газа. Жидкость из сепаратора 18 отделяют от небольшого количества воды в сепараторе 19, объединяют со смесью продуктов после фильтр-пресса 17 и направляют на дальнейшую переработку – отгонку метанола, омыление непревращенных сложных эфиров щелочью при 90оС, отгонку высших спиртов в вакууме от тяжелого остатка (мыла). Метанол возвращают на синтез сложного эфира.
В-34. Окисление. Окислением называют превращение веществ под действием тех или иных окислительных агентов, с образованием кислородсодержащих продуктов. Окисление может быть полное и интереса для нефтехимического синтеза не представляет: C3H8 + 5O2 → 3CO2 + 4H2O.
Неполное окисление может протекать с образованием различных кислородсодержащих продуктов.
1. Без разрыва C-C связи, при котором число УВ-ных связей остается без изменений, к-рое в свою очередь может протекать по насыщенной и по двойной связи. Окисление без деструкции по насыщенной связи имеет место: а) для парафиновых УВ с образованием спиртов и кетонов: RCH2CH3+0,5O2→RCH2CH2OH
RCH2CH3 + O2 → RCOCH3 + H2O; б) при окислении нафтеновых УВ также до спиртов и кетонов. в) для окисления аренов до альдегидов и кислот.
2. Окисление без деструкции по ненасыщенной связи
RCH=CH2 + 0,5 О2 → окись парафина; RCH=CH2 + O2 → RCOCH3 + H2O
3. Окисление с деструкцией – происходит расщепление С-С связи с образованием низкомолекулярных соединений. Окисление имеет место: а) для парафиновых УВ с образованием кислот: RCH2CH3 + 3O2 → R’COOH + CH3COOH + 2H2O
б) при окислении нафтенов с разрушением цикла: циклогексан+2,5 O2→HOOC-CH2-CH2-CH2-CH2-COOH + H2O; в) для ненасыщенных УВ с образованием кислот: RCH=CHR’ + 2O2 → RCOOH + R’COOH
H2C=CH2 + O2 → 2HCHO; г) при окислении ароматики, протекающее с разрушением ароматического кольца.
4. особый вид окисления, сопровождающийся связыванием исходных молекул. а) окислительная конденсация с образованием эфиров: 2RH +1,5O2 → ROOR + H2O ; б) окислительный аммонолиз, протекающий с образованием нитрила: RCH3 + NH3 + 1,5O2 → RCN + 3H2O.
Процессы окисления могут быть гомогенные и гетерогенные. Гетерогенные протекают в паровой фазе на поверхности твердого кат-ра. Также процессы могут быть каталитическими и термическими.
Окислительные агенты.
1. В качестве окислительных агентов используют молекулярный кислород в виде воздуха, технического кислорода и азото-кислородных смесей, где концентрация кислорода ниже чем в воздухе (около 4-5% О2). Технический кислород используют в тех случаях, когда необходима более высокая скорость окисления и когда недопустим унос продуктов окисления с входящим воздухом. Азотно-кислородная смесь используется, когда нужна низкая скорость окисления, при получении нестабильных, способных к дальнейшему окислению продуктов реакции.
2. HNO3 (40-60%). Она является сильным окислителем, исп-ся для деструктивного окисления. Основная проблема при ее использовании в утилизации побочно образующихся оксидов азота, для чего строится установка по переводу их в HNO3.
N2O3 + O2 + H2O → 2HNO3.
3. Пероксидные соединения – H2O2, органические пероксиды ROOH и надкислоты RCOOOH – эти соединения дорогие, взрывоопасные, требуют специальных условий хранения и поэтому используются только в тех случаях, когда невозможно использовать другие агенты.
Техника безопасности и основные трудности в процессах окисления. Все перечисленные окислительные агенты дают с органическими в-вами взрывоопасные смеси, а перекисные соединения являются неустойчивыми. Взрывоопасность определяется пределами взрываемости. В процессах окисления получают развитие побочные реакции: полное окисление, особенно протекающие с большой скоростью при повышении Т. Это приводит к понижению селективности и выхода. Поэтому большое внимание уделяется теплоотводу. При окислении возможно образование широкого спектра кислородсодержащих продуктов, к-рые нужно отделить от целевого и направить на рециркуляцию.
Механизм образования продуктов окисления.
Активными частицами при гомогенном окислении явл. радикалы со свободной валентностью на атоме кислорода или углерода (R˙, ROO˙). Первыми молекулярными продуктами образующимися в зоне реакции, явл-ся гидропероксиды. Образовавшийся тем или иным способом радикал при взаимодействии с кислородом дает гидропероксидный радикал:
R˙ + O2 → ROO˙; ROO˙ + RH → ROOH + R˙ .
При окислении парафин будут образовываться преимущественно вторгидропероксиды с равновероятным расположением гидропероксидной группы при любом из вторичных УВ атомов:
R-CH2-R’ + O2 → R-CH(OO:H)-R’;
при окислении изопарафинов получают третгидропероксиды:
R-CH(R’’)-R’ + O2 → R-C(OOH)(-R’’)-R’;
при окислении алкилароматики гидропероксидная группа занимает α-положение.
Следующие продукты окисления – спирты и кетоны могут образовываться в равных количествах. Образующиеся гидропероксиды в силу их нестабильности взаимодействуют с активными частицами в реакционной массе с образованием спиртов и алкильных радикалов, которые также дают спирты:
ROOH + R’ → ROH + RO’; RO’ + RH → ROH + R’
Кетоны же преимущественно образуются из вторгидропероксидов, которые активизируются за счет отрыва атома водорода от того же углеродного атома, при котором находится пероксидная группа. В результате чего образуется гидропероксидалкильный радикал, который разрушается до кетонов: RCH(OOH)R’ + ROO˙ → → RC˙(OOH)R’ + ROOH → RC(=O)R’ + HO˙
третгидропероксиды образуют спирты и кетоны с деструкцией по схеме: R3COOH + R˙→ R3CO˙ + ROH;R3COOH + R3C˙ → R3COH + R3CO˙; R3CO˙ → RCOR + R˙; R3CO˙ + R3CH → R3COH + R3C
Образовавшиеся системы также способны переходить в кетоны путем отрыва атома водорода от того же атома углерода, при к-ром находится гидроксильная группа через промежуточное образование оксигидропероксидного радикала и оксигидропероксида.
R-CH(-OH)-R’ + R’ → RC˙(OH)-R’+RH + O2→ R-C(OO˙)(OH)-R’+RH → R-C(OOH)(-OH)-R’ + R˙ → RC(=O)R’ + H2O2
Рассмотренный механизм для некаталитических процессов протекает при умеренных Т. В присутствии кат-ра (солей металлов переменной валентности) образование идет не через гидропероксиды, а сразу же из гидропероксидного радикала через промежуточное образование комплексного соединения с металлом, который затем разлагается с переходом металла в высшую валентную форму.
R-CH(OO˙)-R’ + Co(OCOCH3)2 → →
[RCH(R’)-OO˙CO(OCOCH3)2] → RC(=O)R’ + OHCo(OCOCH3)2
При газофазных и термических реакциях в реакционной массе накапливаются преимущественно спирты и альдегиды, образование которых идет также минуя стадию образования гидропероксидов.
Карбоновые кислоты являются продуктами более глубокого окисления и образуются всегда с деструкцией, за исключением боковых цепей аренов:
ArCH3 + O2 → ArCH2OOH → ArCHO → ArCOOH
При окислении парафинов и олефинов наблюдается обязательная деструкция по С-С связи.
RCH2-CH2R’ +2O2 → RCOOH + R’COOH ; При окислении нафтенов происходит образование кислот, сопровождающееся разрушением цикла
бензол + O2 → HOOC-(CH2)4-COOH
Наиболее вероятно образование кислот из кетонов, к-рые активируются за счет отрыва атома водорода от α-углеродного атома по отношению к кетогруппе, с промежуточным образованием кето-гидропероксидного радикала или кетогидропероксида. И тот и другой продукты образуют кислоты с деструкцией.
RCH2-C(=O)-R’+R˙-RH→R˙CH-C(=O)-R’+O2→RC(OO˙)H-C(=O)-R’+RH-R˙→RCH(OOH)-C(=O)-R’
RCH(OO˙)-C(=O)-R’ → RCOOH +R’CO˙; RCH(OOH)-C(=O)-R’ → RCOOH + R’CHO
Кинетика гомогенного окисления. Гомогенное окисление имеет следующие стадии:
1. Зарождение цепи, которое можно вызвать добавлением в реакционную массу инициаторов (для газовой фазы: HNO3,; для жидкой фазы: нестабильные гидропероксиды); воздействием высокой Т: RH+O2→R˙+HOO˙; использованием кат-ров (солей Ме-ов переменной валентности). Стадия зарождения цепи поставляет радикалы только лишь в начальный момент окисления сокращая индукционный период. В стационарном процессе радикалы образуются в результате протекания вырожденного разветвления цепи:
Для газофазной реакции: RCHO + O2 →RC˙=O + HOO˙
Для жидкофазной реакции:
2RCOOH → RCOO˙ + RCO˙ + H2O
Для каталитических процессов:
RCOOH + Mn(OCOCH3)2 → RCO˙ + MnOH(OCOCH3)2
2.Развитие цепи.
3.Обрыв цепи. При газофазных процессах обрыв цепи протекает при столкновении радикалов со стенкой. При жидкофазных процессах наблюдается квадратичный обрыв цепи с образованием молекулярных продуктов. 2RCOO˙ + 2RCOOO˙ → мол. продукты. Обрыв цепи возможен на ингибиторах, в качестве к-рых выступают примеси, содержащиеся в сырье (фенолы, сернистые соединения). Обрыв цепи возможен и на кат-ре, находящемся в нижнем валентном состоянии ROOO˙ + Cu → ROOO˙ + Cu.
Кинетика термического окисления:
1. Зарождение цепи: r0 RH+O2→R˙+HOO˙ (1)
2.Развитие цепи: r1 R˙+O2→ROO˙ (2)
3. Продолжение цепи: r2 ROO˙+RH→ROOH+R˙ (3)
4.Реакция вырожденного разветвления цепи:
rвр ROOH→RO˙+HO˙; 2ROOH→RO˙+ROO˙+H2O (4)
5. Реакция обрыва цепи: rt 2ROO˙→ мол. пр-ы.
Запишем уравнение скорости реакции окисления (3)
r2=K2[ROO˙][RH]
Используя условие стационарности, запишем уравнение для скорости обрыва цепи и выражд. развет. цепи. rt=Kt[ROO˙]2 ; rвр=Kвр[ROOH]n;
[ROO˙]=(Kвр/Кt)1/2 [ROOH]n/2 ;
rt =K2 (Kвр/Кt)1/2[RH][ROOH]n/2
Это выражение для скорости термического окисления имеет место для случая избытка кислорода. При недостатке кислорода лимитирующей будет реакция 2, а обрыв цепи будет идти по алкильным радикалам. r1=K1[R˙][O2];
r1=K1(Kвр/Кt)1/2[O2][ROOH]n/2
Последовательные реакции дают целый ряд кислородных соединений, побочных продуктов основным способом их снижения является регулирование степени конверсии.
По степени конверсии процессы можно разделить:
1.Получение стабильных, стойких к дальнейшему окислению продуктов. Когда степень конверсии не играет существенной роли (получение кислот и ангидридов). Селективность допускается 95 – 99%.
2.Получение нестабильных веществ способных разрушаться или окисляться более глубоко (получение спиртов, гидропероксидов и кетонов). Селективность 5 – 30%.
Энергия активации полного окисления очень высока. Поэтому с повышением Т получается развитие полного окисления. Для его предотвращения необходимо уделять внимание теплоотводу. На селективность также влияет турбулизация, поэтому вводимый окислительный агент распространяется по всему реакционному объёму с помощью барбатирующих устройств.
Реакторы жидкофазного окисления. Используют колонные аппараты Н=10 –15 м, D =2 – 3 м. Выполненные из нержавеющей стали и цветного металла. Давление для жидкофазного процесса влияния на скорость не оказывает и поддерживается 0,1-8 МПа. Для создания жидкой фазы температурный режим 50 – 2500 С.
1. Окислительная колонна противоточная, периодичного действия с выносным холодильником и расширенной верхней частью, откуда выводится воздух, обедненный кислородом с примесями летучих продуктов и сырья. С низа выводятся продукты окисления. Применяется для низкоэкзотермических процессов.
2.Прямоточная колонна с внутренним охлаждением используется для высокоэкзотермических процессов с высокой степенью конверсии.
3. Каскад ректоров с последовательной подачей исходного реагента и параллельной подачей воздуха. Тепло реакции снимается испарением непревращённого сырья выносимого вместе с воздухом. Используется для получения нестабильных веществ для получения суммарной степени конверсии и сохранения селективности.
4.Секционированная колонна в которых за счёт секций устраняется отрицательный эффект продольного перемешивания, предотвращается развитие последовательных побочных реакций. Используются для высокоэкзотермических процессов с невысокой конверсией.
В-35. Производство оксида этилена.
СН2- СН2
\ / Ткип=10,7˚С; пределы взрываемости 3-80%
О Используется в след. напр-ях:
1) для пр-ва гликолей: С2Н4О +Н2О→НОСН2-СН2ОН
2) пр-во акрилонитрила: НСN + C2Н4О → СН3-СН2СN + Н2О
3) в пр-ве ПАВ путем оксиэтилирования ВЖС, СЖК и диамина. Так получают ПАВ неионногенного типа с низким пенообразованием.
4) пр-во этаноламинов: NH3+ С2Н4О→ НО-СН2-СН2-NН2
Оксид этилена можно получать хлорным методом путем хлоргидратации хлорэтилена с последующей обработкой гидроксидом кальция:
СН2=СН2+ Сl2+Н2О → ClCН2-СН2ОН ClCН2-СН2ОН+ + Са(ОН)2 → С2Н4О +СаСl2+ Н2О
Этот метод имеет недостатки:
1) использование Сl2;
2) многостадийный пр-сс;
3) использование Са(ОН)2;
4) получение большого кол-ва сточных вод.
Поэтому используют прямое окисление этилена:
СН2=СН2 + 0,5О2 → С2Н4О → СО2 + СО
Q= 117 кДж/моль
СН2=СН2 → СО2 + СО Q= 1410 кДж/моль
Для этой реакции характерно развитие паралл. и последоват. реакций.
ЕС2Н4О= 63 кДж/моль; ЕСО2= 84 кДж/моль , следовательно, развитие побочных р-ий приводит к повышенному выходу СО2 вследствие повышения t˚C. Зависимость ск-ти обр-я продуктов р-ии от t˚C.
Кроме t˚C на селективность оказывает влияние ст. конверсии и время контакта, кот. составляет 1-4 сек. За один проход реактора ст. превращ.= 35%, суммарная = 40-70%, kt подбирается таким образом, чтобы при достаточно низкой t˚C обеспечить высокую ск-ть
р-ии. Таким kt явл-ся серебро. Оно активно при 220˚C и селективно до 280˚C, т.к. это дорогой kt его используют на носителе с добавкой промотора – дихлорэтан, такая добавка повышает селективность на 5%, носителем яв-ся корунд. Kt служит 6-9 лет.
Процесс ок-ия можно проводить при недостатке этилена, тогда скор-ть р-ии будет опр-ся абсорбцией прод-ов р-ии, но и конц-ей этилена.
 , b – абсорбционный
коэф-т.
, b – абсорбционный
коэф-т.
В случае избытка этилена скор-ть опр-ся абсорбцией прод-ов р-ии и конц. О2

механизм действия kt основан на хемосорбции. Первонач-но сорбируется О2, а затем на ок-ой поверх-ти kt сорбируется этилен, он взаимодействует с ок-ой поверх-ю с обр-ем прод-ов р-ии. В прмышленности реализовано два способа ок-ия этилена:
1) ок-ие воздухом; 2) ок-ие техническим О2.
Технология ок-ия этилена воздухом. Процесс осущ-ся в двух реакторах расположенных последовательно, извлечение оксида этилена осущ-ся после каждого реактора этим достигается более высокая селективность при достаточно высокой ст. конверсии. Кроме этого осущ-ся рециркуляция газа на 1ой ступени, что даёт полное исп-ие этиленом кислорода с понижением взрывоопасности.
Воздух, этилен и рециркулят смешиваются с получением состава: 4-6% С2Н4, 6-8% О2, 8-10% СО2, остальное азот. Ок-ие осущ-ся в трубчатом реакторе 1, где давление поддерживают 2 МПа с целью сокращения объёма аппаратуры, тепло р-ии снимается водой с получением пара среднего и низкого давления. Продукты р-ии охлаждаются в теплообменнике 2 рециркулятом и поступают на абсорбцию 1ой ступени в обсорбер 3 в который подаётся вода. Содержание оксида этилена в продуктах р-ии до 1,5% (т.к. оксид этилена очень хорошо растворяется в воде именно в этом соотношении). Абгазы выходящие сверху частично возвращаются в реактор 1, а остальная часть на 2ую ступень окисления в реактор 4, предварительно подогреваясь в теплообменнике 5 отходящими продуктами. Из реактора 4 реакционная смесь поступает на 2ую ступень извлечения водой. Водные растворы оксида этилена из абсорберов 3 и 6 объединяются и поступают на отпарку. По данной технологии общий выход оксида этилена составляет 60%.
В-36. Жидкофазное окисление парафинов.
Гомогенное окисление парафинов возможно по следующим направлениям:
1. Окисление в газовой фазе с целью получения спиртов и альдегидов.
2. Термическое окисление парафинов в жидкой фазе в присутствии борной кислоты, с целью получения высших спиртов.
3. Каталитическое окисление в жидкой фазе с получением карбкислот.
Способность различных низших парафинов окислятся возрастает с удлинением цепи.
Наибольшее распространение нашло окисление пропилена в результате чего получается смесь спиртов и альдегидов, окисление протекает с деструкцией.
C3H8 + O2 → CH3CH2CH2OH + O2 → CH3CH2CHO
C3H8 + O2 → CH3CH(OH)CH3 + O2 → CH3COCH3
C3H8 + O2 → CH3OH + CH3CHO
C3H8 + O2 → C2H5OH + HCHO
Процессы осуществляются при Р=0,7-1 МПа Т=430-450оС, подаваемая смесь содержит рециркулят, свободный УВ и воздух в соотношении 7:1:2. Процесс осуществляется в адиабатическом трубчатом реакторе, где Т поддерживается за счет тепла реакции. Реакционные газы проходят змеевиковый разлагатель, заполненный керамической насадкой для разрушения гидропероксидов. Далее смесь поступает абсорбер, к-рый орошается раствором формальдегида (12-14%), где происходит закалка и поглощение преимущественно спиртов. Далее смесь поступает на вторую ступень абсорбции для извлечения оставшихся О-сод. веществ. Газовый поток со второй ступени возвращается на рециркуляцию, а низшие потоки из абсорберов направляются на разделение.
Окисление н-парафинов в спирты происходит в отсутствии кат-ра и в жидкой фазе.
Способы получения ВЖС: восстановление кашалотового жира; восстановление метиловых эфиров СЖК; выделение ВЖС в производстве СЖК; прямое окисление парафинов молекулярным кислородом, получаются преимущественно вторичные спирты.
Пути использования ВЖС:
1) в производстве ПАВ;
2) в синтезе депрессорных присадок;
3) как растворители;
4) в бумажной, текстильной и кожевенной промышленности;
5) для производства пластификаторов.
Катализаторы окисления парафинов до кислот. Окисление в карбоновые кислоты идет с деструкцией, возможны два варианта окисления низших парафинов в уксусную кислоту и окисления высших парафинов в СЖК. Окисление низших парафинов в уксусную кислоту.
Основные закономерности и параметры процесса
1. Влияние температуры. Окисление парафина уже возможно при 100°С, но оно протекает очень медленно. Скорость окисления увеличивается с повышением Т (при 160°С время окисления 20-25 часов), но при такой Т образуется большое кол-во продуктов деструкции. Использование кат-ра позволяет проводить процесс с достаточно высокой селективностью при невысоких Т, которая в начальный момент составляет 125-130°С для формирования гомогенного катализатора. Затем происходит постепенное снижение со скоростью от 2 до 5°С в час до Т=105-110°С. При таком переменном температурном режиме время окисления сокращается до 15-20 ч. Выход кислот 80%.
2. Влияние давления. Скорость окисления возрастает с повышением давления до 0,2 МПа; при более высоком Р наблюдается ухудшение качества продукта в той степени, в какой повышается растворимость воздуха в парафине.
3. Влияние кат-ра. В качестве кат-ра используются Na-Mn или K-Mn, компонентное соотношение 1:1 (Na:Mn; K:Mn). В качестве натриевого компонента используются соли жирных кислот RCOONa. В качестве Mn компонента исп-ся либо оксиды марганца, либо марганцевые соли жирных кислот (MnO2,(RCOO)2Mn). Лучше использовать KMnO4.
Активной формой кат-ра является его высшая валентная форма. В процессе окисления марганец восстанавливается до низшей валентности, раствор обесцвечивается, т. к. Mn(2+) способен выпадать в осадок. Для предотвращения выпадения в осадок необходим натриевый или калиевый компонент. Марганец катализирует зарождение цепи, снижая индукционный период и катализирует разложение гидроперекиси. При увеличении концентрации марганца происходит снижение выхода кислот вследствие развития обрыва цепи.
4. Влияние скорости подачи воздуха. Обычно она составляет 60-70 м3 на одну тонну парафина. Это соответствует линейной скорости 0,1 м/с. Избыток воздуха приводит к увеличению теплового эффекта, а следовательно к перегреву и деструкции. С избытком воздуха уносится часть продукта.
5. Влияние времени окисления и глубины окисления. Скорость накопления продуктов окисления регистрируется функциональным числом. Накопление кетонов идет через максимум, что говорит о дальнейшем их превращении в кислоты, а увеличение выхода кислот наблюдается до времени окисления 15-20 часов. Оптимальными условиями считаются время окисления 15-20 часов, Р=0,2 МПа, скорость подачи воздуха 0,1 м/с, переменный температурный режим.
Технология окисления нефтяного парафина в СЖК.
Исходный парафин смешивается с кат-ром и рециркулятом в емкости 1. Соотношение свежего парафина и рециркулята 1:2 (окислительная шихта). Окислительная шихта подается в полый реактор 2, снабженный распределительным устройством для подачи воздуха и рубашкой для подогрева и охлаждения. Отработанный воздух сверху выводится на промывку в скруббер 3, затем отправляется на дожигание в печь 4 и затем сбрасывается в атмосферу.
Оксидат с низа колонны 2 поступает в шламоотделитель 5 и затем поступает в промывную колонну 6 для удаления водорастворимых низших кислот. Смесь кислот и др. кислородсодержащих продуктов поступает в омылитель 7, где происходит взаимодействие с водным раствором соды. В результате свободные кислоты переводятся в мыло. Омыление необходимо, чтобы предотвратить образующиеся кислоты при их выделении от деструкции и дальнейшего окисления, увеличить расход кислот, т. к. на стадии омыления переводятся в мыло не только свободные кислоты, но и другие кислородсодержащие продукты. В аппарате 8 подвергаются омылению эфиры водным раствором щелочи. Реакционная смесь подогревается в теплообменнике 9 и поступает в автоклав 10, где происходит выделение неомыляемых продуктов. Они возвращаются в узел приготовления окислительной шихты. Т= 180°С, Р= 2 МПа. Омыление оставшихся кислородсодержащих продуктов завершается в трубчатой печи 11 при более жестких условиях (380°С; 2,5 МПа). Здесь переводят в мыло кетоны и лактоны. Далее смесь проходит дросселирующий клапан, в результате чего в сепараторе 12 выделяется мыло и УВ-часть отдает свое тепло в теплообменнике 9. В сепараторе 12 из нее выделяется вода, а сами они в виде 2-х неомыляемых продуктов выводятся для последующего их выделения с последующим гидрированием. Мыло подается шнеком, добавляя воду для получения мыльного клея. Мыльный клей поступает на разложение серной кислотой в аппарат 15. В аппарате 16 от СЖК отделяют Na2SO4, а СЖК отправляют на вакуумную ректификацию для разделения по фракциям.
В-37. Жидкофазное окисление аренов.
Осн. реакциям являются:
1. Окисление алкил бензолов до гидропероксидов с последующим их кислотным разложением на фенол и кетоны:
С6Н5СН(СН3)2+O2→С6Н5С(ООН)(СН3)2+H+→С6Н5ОН+(СН3)2СО
2.Окисление метилбензола в кислоту С6Н5СН3+О→С6Н5СНО→ С6Н5СООН
Рассмотрим 1. пример:
Окислению в гидропероксиды подвергают преимущественно кумол и реже изопропилнафталин и диизопропилбензол. Этот метод лег в основу кумольного метода получения фенола и ацетона. Процесс идет по свободно-радикаьному механизму в отсутствии кат-ра, т.к. они разлагают образовавшиеся гидропероксиды. Для сокращения индукционного периода в исходную смесь добавляют 3-4% гидропероксида окисляемого УВ. В связи с этим механизм можно представить так:
С6Н5СН(СН3)2+ROO• →С6Н5C•(СН3)2+ ROOH +О2→ С6Н5СОО•(СН3)2→С6Н5СОСН3+СН3О•
+RH|___________→С6Н5С(ОOH)(СН3)2 + R•
Присоединение гидропероксидной группы происходит по третичному УВ-ному атому с образованием побочных продуктов (ацетофенон и фенилкарбинол) С6Н5С(ООН)(СН3)2+С6Н5С(СН3)2→
→С6Н5С(ОН)(СН3)2 +С6Н5С (О•)(СН3)2
Разложение осуществляется в присутствии кат-ра кислотного типа при Т=40-60°С. Механизм сложно-ионный. Первоначально протонируется молекула гидропероксида, затем отщепляется вода, происходит миграция кислорода к фенольной группе, затем происходит присоединение воды; внутримолекулярное перемещение зарядов и завершается процесс деструкции с образованием фенола и ацетона и регенерацией протона водорода.
С6Н5С(СООН)(СН3)2+H+↔С6Н5С(ООН+2)(СН3)2+Н2О↔С6Н5С(О)(СН3)2→С6Н5ОС+(СН3)2+Н2О→
→С6Н5ОС(ОН2+)(СН3)2→С6Н5О+(Н)С(ОН)(СН3)2→ →С6Н5ОН+ (СН3)2СО+Н+
Разложение гидроперекиси может осуществляться в различных реакционных узлах:

1.Прямоточная циркуляционная система
Процесс экзотермичный выделяющееся тепло снимается водой. Этот процесс неперспективный и имеет ряд недостатков: низкая доля полезного объема , в следствие рециркуляции высокое время реакции, дающее образование побочных продуктов, высокая металлоемкость.
2. Наиболее перспективный горизонтальный аппарат с секционированными перегородками
Тепло снимается ацетоном
Тех. схема кумольного метода получения фенола и ацетона.
Технология состоит из 3-х стадий: 1.Алкилирование бензола пропиленом; 2. Окисление; 3.Разложение.
Окисление кумола проводят воздухом в секционированной колонне1, в к-ой установлены змеевики для поддержания переменного теплового режима по высоте колонны. В верхнюю часть колонны подается сырье с Т=120°С с постепенным понижением Т до 105-110°С. Т.к. происходит увеличение содержания гидроперекиси, а следовательно и возможности ее деструкции. Окислительная шихта готовиться в аппарате 5, куда подается свежий и непревращенный ИПБ и гидроперекись. Шихта подогревается в теплообменнике 6 и идет в верхнюю часть К-1. С воздухом обененным О2 носится часть непревращенного ИПБ и легкие побочные продукты. Они охлаждаются в холодильнике 2. Конденсат отделяется в сепараторе 3 и стекает в прмывную колнну 4. откуда сверху углеводородный слой поступает в емкость приготовления шихты 5, а вода идет в стоки. Оксидат содержащий 28-30 % гидропероксида охлаждается исходной смесью в теплообменнике 6. затем сбрасывается Р до 4 кПа и поступает в узел укрепления 7. Там происходит отгонка неревращенного ИПБ, к-ый отправляется на промывку в К-4. Узел укрепления состоит из 2-х последовательно работающих насадочных колонн. В первой из них Р=4кПа, гидроперекись укрепляется до 70-75%; во второй колонне Р=600-605Па гидроперекись укрепляется до 88-92%. Укрепленная гидроперекись идет в узел кислотного разложения 8, работающего по одному из указанных методов. Далее продукты разложения обрабатываются щелочью, образующиеся сульфаты выделяются в сепараторе 9.Далее продукты разложения идут в К-10, где происходит разложение ацетонового и фенольного потоков, каждый из которых проходит систему колонн для получения товарных продуктов. В случае выделения фенола легкими примесями является ИПБ и ацетофенон, а тяжелыми- различные смолы. Помимо фенола и ацетона м/о получить ряд др. продуктов. Например, двухатомные фенолы. (гидрохинон и резорцин).
В-38. Гетерогенно-каталитическое окисление.
Гетерогенно-каталитическое окисление имеет место при синтезе следующих продуктов:
1. Получение акролеина, метакролеина и их кислот. Окисление осуществляют по насыщенному атому углерода олефинов. При этом двойная связь сохраняется. Например, окисление пропилена в акролеин и акриловую кислоту.
СН2=СН-СН3 + О2 – H2O → СН2=СН-СНО + 0,5О2 → → СН2=СН-СООН
2.Синтез акрилонитрила окислительным аммонолизом, где происходит взаимодействие олефинов с аммиаком в присутствии кислорода
RCH3 + NH3 +1,5O2 → RCN + 3 Н2О ; R: CH2=CH-
3.Окисление аренов с образованием ангидридов ди- и тетрокарбоновых кислот.

4. Синтез α-оксидов окислением олефинов.
СН2=СН2 + 0,5О2 → α-оксид
Кат-ры гетерогенно-каталитического окисления.
1. металлы (Ag, Cu);
2. Оксиды металлов ( CuO, Cu2O, V2O5);
3. Сложные катализаторы ( ванадаты, вальфроматы) - представляют смесь оксидов, иногда солей: ZnO* V2O5*CoWO3. Кат-ры могут использоваться в различных формах. Без носителя используются сложные и оксидные кат-ры. Носитель-пемза, Al2O3.
Механизм гетерогенно-каталитического окисления.
В основу катализа положена хемосорбция реагента на поверхности кат-ра. Рассмотрим сорбцию О2: Кислород сорбируется на поверхности металла как с диссоциацией, так и без диссоциации. Необходимый для связи электрон поставляет Ме и Кат. переходит в состояние ион-радикала Ag + О2 → Ag-O-O˙- + Ag → 2Ag-O˙-
На поверхности оксидного и солевого кат-ра кислород сорбируется слабо. При этом сорбция происходит по иону переходного Ме, к-й переходит в высшее валентное состояние.
Реакторы гетерогеннокаталитического окисления.
УВ с О2 образуют взрывоопасные смеси, для избежания взрыва используют следующие приемы:
1) Окисление рециркулирующими газами с добавкой свежего воздуха или О2 при низкой концентрации УВ (3-5%).
2) Использование избытка УВ при небольшом содержании воздуха или технического кислорода.
3) Разбавление водяным паром.
Ввиду высокой экзотермичности процессов нашли применение следующим конструкциям реакторов:

(а) Кожухотрубный р-р. Используется в том случае, когда применяется дорогой kаt и в реакциях с псевдоожиженным слоем kаt будет уносится «–» – высокая металлоемкость, низкая доля полезного объема, невозможность создания изотермического режима.
(б) Несекционированный р-р. С псевдоожиженным слоем kаt с внутренним змеевиком и батареями циклонов в верхней части для улавливания частиц Каt .
(в) Секционированный р-р. с псевдоож-ным слоем kаt. Меньше продольное перемешивание из-за секций.
(г) Реактор с восходящим потоком кат-ра, продукты вместе с kаt поднимаются по восходящей трубе; там они охлаждаются водным конденсатом, затем Каt отделяется от продуктов в бункере или сепараторе и возвращается в нижнюю часть р-ра (дозатор).
Производство малеинового ангидрида. В промышленности его получают преимущественно парофазным окислением бензола, бутенов или бутанов.
1)
![]()
2)
.![]()
Тех. схема парофазного окисления бензола в малеиновый ангидрид.
Бензол смешивается с воздухом, который подается в 25-30-кратном избытке, создавая концентрацию бензола в смеси 0,6-1,6%. Далее бензоло-воздушная смесь подогревается отходящими продуктами реакции в теплообменнике 1 и далее поступает в трубчатый реактор 2, заполоненный kat. Тепло реакции утилизируется расплавом солей с выработкой пара высокого давления, далее смесь охлаждается в холод-ке 3, с выработкой пара низкого давления и после теплообменника 1 и холод-ка 4 поступает в узел выделения малеинового ангидрида 5, оформленного в виде ребристых конденсаторов или в виде сепараторов. Сконденсировавшиеся малеиновый ангидрид собирается в емкости 6, а не уловленный поступает в абсорбер 7 на улавливание водой с получением 40% р-ра малеиновой кислоты, сверху абгазы уходят на сжигание и далее в атмосферу. Малеиновая кислота поступают в отпарную К-8 на дегидрирование, малеиновый ангидрид с куба К-8 идет в сборник 6, откуда он направляется на заключительную очистку от примеси в К-9.
В-39. Синтезы на основе оксида углерода
Основаны на использовании синтез-газа, широко распространенны, т.к. в этих синтезах используется дешевое сырье. Они реализованы в следующих направлениях: 1) Синтезы непосредственно из CO и H2 с получением смеси УВ и O-содержащих соединений. 2) Процессы оксосинтеза или гидроформилирования, процессы карбоксилирования, где помимо синтез газа используются олефины.
Рассмотрим 1) направление:
1. Синтез смеси УВ, к-ые получают на Ni кaт-ре при Т>200°C. Этот процесс впервые был разработан Фишером и Тропшем в 1923г. CO + 3H2 ↔ CH4 + H2O
На оксиде железа при P=3МПа была синтезирована смесь УВ, называемая когазином.
2nCO + (n+1)H2 → CnH2n+2 + nCO2
До разработки нефтяных месторождений этот процесс широко использовался для получения синтетического бензина, выделяемого из когазина в виде узкой бензиновой фракции.
2. В 1924г. Патаром при P=10-15МПа на ZnO впервые были синтезированы О-содержащие соединения из синтез-газа при Т=250-300°С, из к-ых наибольшую ценность представляет CH3OH. CO + 2H2 ↔ CH3OH;
С тех пор этот метод получения метанола явл-ся единственным в промышленности.
Производство метанола.
Метанол явл-ся многотонажным продуктом и используется, как в виде целевого, так и промежуточного продукта. В виде целевого используется, как растворитель или высокооктановая добавка. В виде промежуточного, как метилирующий агент в производстве метиловых эфиров и метиламинов, в производстве формальдегида и МТБЭ. Ткип(CH3OH)=64,7°С,
пределы взрываемости 6-34,7%. Синтез CH3OH протекает по обратимой экзотерм-ой реакции, выделение тепла составляет 110,8 кДж/моль. Синтез идет с уменьшением объема, поэтому для сдвига равновесия вправо нужно увеличить давление до5-40МПа, в зависимости от используемой Т. Т может быть 350-400°С, но при всех Т повышенное давление благоприятно сказывается на выходе метанола. Долгое время в качестве кат-ра использовалась смесь ZnO и CrO в соотношении 8 : 1. Механизм действия такого кат-ра аналогичен процессам гидрирования и заключается в сорбции исходных реагентов на пов-ти кат-ра с участием электронных переходов при последующем взаимодействии хемосорбционных центров с образованием продукта.
K + CO↔K:::C=O + H2↔K:::CH-OH + H2↔K + CH3OH
Хемосорбированные частицы имеют св-ва радикала.
Реакционные узлы синтеза метанола

а) Трубчатый реактор, где тепло отходящих продуктов исп-ся для подогрева исходных реагентов;
б) Адиабатический реактор секционированный, где на решетках установлены перфорированные корзины заполненные кат-ра. Тепло реакции снимается за счет подачи между слоями кат-ра синтез-газа. Это самая распространенная конструкция, к-ая позволяет вести процесс в изотермич-ком режиме без перегрева Ф=95%;
в) Представляет собой трехфазную систему, в к-ой осуществляется синтез в инертном растворителе, на суспензированном в нем гетерогенном кат-ре. Через эту суспензию барботируется синтез-газ. Такая система позволяет доводить конверсию до 35%, использовать синтез-газ меньшей чистоты и вести процесс в изотермическом режиме.
Технология производства метанола.
В современных установках обычно совмещается установка получения синтез-газа с установкой получения метанола. УВ-ный газ сжимается компрессором 1 и поступает в конвертор 2 для поучения СО и Н2. Т.к. Т в конверторе высокая, синтез-газ охлаждается в котле утилизаторе 3 с выработкой пара высокого давления, далее в холодильнике 4 и сжимается компрессором 5 до рабочего давления. Далее газ идет в адсорбер 7 для удаления из синтез-газа пентакарбонила железа Fe(CO)5, к-ый образуется при взаимодействии СО с трубами при высоком давлении и разлагается, образуя мелкодисперсное железо, к-ое катализирует побочные реакции. Далее синтез-газ делится на два потока: холодный подается между слоями кат-ра в реактор 8, а другой поток подогревается отходящими продуктами реакции в теплообменнике 9 и также поступает в реактор 8. Получаемые продукты реакции на выходе из реактора также делятся на два потока: один поток греет исходное сырье, тепло второго потока используется в котле-утилизаторе 10 для выработки пара. Далее продуктовые потоки объединяются, охлаждаются в холодильнике 11, в сепараторе 12 происходит разделение продуктов и синтез газа.
Далее синтез-газ дожимается компрессором 6 и возвращается в процесс. Продукты реакции из сепаратора 12 дросселируются и поступают в колонну 13, где при давлении, близком к атмосферному, отгоняются первоначально легкие фракции, а в колонне 14 из CH3OH отделяют тяжелые примеси. Выход CH3OH = 90-95%.
В-40. Процессы гидроформилирования.
Это процесс взаимодействия синтез-газа с олефинами, где происходит присоединение по двойной связи атома С и формильной группы с другой стороны.
Чаще получают следующие спирты: С4 – производство растворителя; С7-9 – для производства пластификаторов; С10-18 – для произ-ва ПАВ.
Для получения ROH используют олефин с числом атомов С на 1 меньше, чем у получаемых ROH.
Введение молекулы кислорода в молекулу олефина проводят с помощью реакции с одним из трех доступных реагентов: H2O, O2 и CO. Реакции протекают в присутствии kat-ов (карбонилов переходных Ме). При использовании α-оолефина образуются альдегиды, как н-, так и изо-строения. В качестве главного продукта образуются н-альдегиды.
Промышленное применение процессов гидрофор-ия.
1) Получение н-масляного ангидрида из пропилена и синтез-газа.
2) Синтез высших спиртов из α- олефнов ( С8 и выше). 3)Производство пропионового альдегида из пропилена. Гидрированием пропионового альдегида получают пропионовый спирт, а окислением – пропионовую к-ту.
Кат-ры: гидрокарбонилы переходных Ме, активность которых уменьшается в ряду: Rh>Co>Mg>Fe>Cn>Mo>W>Ni. Видно, что активность Rh и Со выше, поэтому гидрофор-ие катализируется растворимыми карбонилами Со и родия (НСо(СО)4). Кат-р генерируется из тонкоизмельченного Ме или солей с использованием синтез газа при повышенной Т и Р.
Механизм гидрофор-ия на примере этилена.
1). Диссоциация кат-ра с образованием поляризационно – ненасыщенного комплекса НСо(СО)4↔НСо•(СО)3 + СО
2). Внедрение этилена по связи Со-Н.
НСо•(СО)3 + СН2=СН2 → НСо•(СО)3↔
↔ СН3СН2 Со•(СО)3 ↑
СН2=СН2
3). Происходит внедрение по связи Со-С с образованием алкилацилкарбонилСо, к-рый при взаимодействии с Н2 или с Кат образует альдегиды- продукты реакции.
СН3СН2Со•(СО)3++СО↔СН3СН2Со(СО)4↔
↔СН3СН2 СОСо(СО)3→ Ι, ΙΙ
Ι +H2→СН3СН2СОСо(Н2)(СО)3→
→СН3СН2СНО + НСо•(СО)3
ΙΙ +НСо(СО)4 →СН3СН2СНО + Со2(СО)7
4). Из Со2(СО)7 регенерируется кат-тор:
Со2(СО)7 + СО↔ Со2(СО)8 + Н2 ↔ 2НСо(СО)4.
Реакционные узлы. Оформление определяется: способом приготовления кат-ра; способом его выведения из продуктов реакции. Способы выделения кат-ров из продуктов реакции: - понижение Р; - химический способ: заключается в обработке продуктов реакции Н2SО4 и Н2О2 с разрушением карбонилов Со.
Со2(СО)8 + 2 Н2SО4 + 2 Н2О2 → 2Со SО4 + 4Н2О + 8СО Далее сульфат Со переводят в активные гидрокарбонилы Со - обычная отгонка при использовании стабильных устойчивых кат-ров.
ТРИАДНАЯ СХЕМА. В катализере на насадке находится Ме- ий Со. При пропускании через к-ый синтез – газа образуется активный гидрокарбонилы Со, растворимые в у/в или в случае низших олефинов – в растворителе. Активная форма кат-ра из катализера подается в нижнюю часть реактора, куда подается оставшийся синтез – газ. В реакторе протекает реация гидроформилирования, которая является экзотермичной и необратимой. В реакторе установлены змеевики для снятия тепла от продуктов реакции. В сепараторе отделяется синтез – газ, к-ый поступает на рециркуляцию, а продукты реакции проходят через дроссель для понижения Р ; при том происходит разрушение карбонилов и гидрокарбонилов Со. В декатализере выдуваются подукты реакции, а Ме-ий Со осаждается на насадке. По мере накопления Со катализер и декатализер меняются местами.
ЭКСТРАКЦИОННО - СОЛЕВАЯ.
В карбонилобразователе 1 происходит образование активной формы Со-го кат-ра при пропускании синтез – газа через Со-ую соль, поступающую из экстракционного узла 3. В реактор 2 подается оставшаяся часть синтез – газа, олефин и раствор Кат-ра из аппарата 1. В сепараторе выделяются непревращенный синтез – газ. А в экстракционном узле 3 кат-р отделяется от продуктов реакции с помощью химических реагентов. Выделившийся СоSО4 здесь же взаимодействуют с подоваемой натриевой солью с образованием нафтоната Со, к-ый возвращается в аппарат 1, а Na2SO4 отводится. ИСПАРИТЕЛЬНО – СОЛЕВАЯ.
В карбонилобразователе 1 образуется активная форма кат-ра при пропускании синтез–газа через Со-ую соль, поступающую из куба колонны 4. В реакторе 2 протекает реакция, куда поступает кат-р, синтез–газ, олефин. В сепараторе отделяется непревращеннный синтез- газ . Продукты реакции дросселируются, кат-р разрушается и выделившийся Ме-ий Со взаимодействует с кислотой, образующейся при окислении альдегида воздухом в аппарате 3. В колонне 4 из продуктов реакции выделяется Со-вая соль, к-ая возвращается в карбонилобразователь 1. ИСПАРИТЕЛЬНАЯ. Данный узел используется в случае стабильного кат-ра, который отделяется от продуктов реакции в колонне 2 и возвращается в реактор.
В-41.Процессы конденсации по карбонильной группе.
Это реакции альдегидов и кетонов с различными реагентами. Главное их назночении состоит в получении изопрена, капролактама и пентаэритрита.
Классификация:
1. По типу и количеству стадий протекания реакций
а).одностадийное присоединение по карбонильной группе RR`CO + HCN ↔ RR`C(ОН)(CN)
б).двухстадийное последовательное присоединение реагента с промежкуточным образованием ацеталей и последующей их дегидротацией
RR`CO + ROH↔ RR`C(ОН)(OR) ↔ RR`C(OR)(OR) + H2O
в).однастадийное присоединение азотистых оснований с внутримолекулярной дегидротацией
RR`CO + NH2X ↔ RR`C=NX + H2О
2.По типу присоединяющегося реагента
а).Реакции альдегидов и кетонов с основаними. В этом случае используется кат-р кислотного типа, активирующий альдегид или кетон путем протонирования атома кислорода с образованием карб – катиона, к-ый способен взаимодействовать с реагентом.
RCНО RCН(ОН+)→RC+НОН
б).Реакции альдегидов и кетонов со слабыми кислотами. В этом случае используется кат-р основного типа активирующий молекулу реагента путем перевода ее в сопряженное основание, к-ое является сильным нуклеофилом. НО- + НА→ А- + Н2О
Нуклеофил способен взаимодействовать с нуклеофилом или кетоном с образованием промежуточного продукта, к-ый взаимодействует с реагентом с образованием продуктов р-ции и регенерации нуклеофила.