

Lectures.Part.2
.1.pdf21
Залежність швидкості реакції від температури
Як відомо, швидкість величезної більшості хімічних реакцій стрімко зростає із збіль-
шенням температури. Саме тому нагрівання є дуже поширеним прийомом як у хімічній практи-
ці, так і в інших галузях: техніці, кулінарії, побуті і фармації зокрема. З цим явищем ми стикає-
мось і в біології: при зниженні температури усі процеси в деяких організмах суттєво сповіль-
нюються і вони переходять у стан анабіозу.
За законом діючих мас:
w |
dcA |
kc p cq |
cr |
|
|||
|
d |
A B |
c |
|
|
|
Отже, вплив температури може здійснюватись через зміну концентрації, через порядок реакції і через зміну константи швидкості реакції. З досвіду відомо, що основний вплив температура вчиняє на константу швидкості. Тому, кажучи про вплив температури на швидкість, мають на увазі зміну константи швидкості.
Для багатьох хімічних реакціях, що протікають у інтервалі 0-100 ºС, справедливе емпі-
ричне правило Вант-Гоффа: при підвищенні температури на 10 ºС (10 К) швидкість реакції зро-
стає у 2-4 рази. Відношення швидкості реакції при температурі Т до швидкості при температурі Т+10 називають температурним коефіцієнтом Вант-Гоффа γ.
|
|
w |
10 |
|
|
k |
T 10 |
|
|
|
1 2 |
T |
|
|
|
|||||||||
|
|
T |
|
|
|
|
|
|
|
(31) |
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
w |
|
|
|
|
|
k |
T |
|
|
|
1 2T 10 |
|
||||||||||
|
|
|
T |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
У випадку, коли зміна температури не рівна 10 ºС, зручно користуватися рівнянням: |
|
|||||||||||||||||||||||
|
w |
|
|
|
k |
T2 |
|
|
|
1 2 |
T1 |
|
|
|
T2 T1 |
|
|
|||||||
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
T2 |
|
|
|
|
|
|
|
|
|
|
|
10 |
|
(31) |
||||||||
|
w |
|
|
k |
|
|
|
|
1 2T2 |
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
T1 |
|
|
|
|
T1 |
|
|
|
|
|
|
|
|
|
|
|||||||
Правило Ван-Гоффа наближене і не враховує залежність температурного коефіцієнта від температури, оскільки, , при зростанні температури, прямує до одиниці.
Голландський учений Арреніус експериментально встановив більш точну залежність константи швидкості від температури:
ln k A |
B |
(32) |
|
T |
|||
|
|
В сучасному вигляді рівняння Арреніуса (32) представляють:
ln k ln k |
|
|
E |
, |
(33) |
||
0 |
RT |
||||||
|
|
|
|
|
|||
|
|
|
|
|
|
||
|
|
|
або: |
|
|
||
k k |
e E RT |
|
(34) |
||||
0 |
|
|
|
|
|
|
|
© Ю.Павловський 2011
|
|
|
22 |
|
Або у диференційній формі: |
|
|
|
|
|
d ln k |
|
E |
(35) |
|
dT |
RT 2 |
||
|
|
|
||
Коефіцієнт рівняння k0 - передекспоненційний множник, має розмірність константи швидкості відповідного порядку. Для елементарних реакцій k0 можна розрахувати теоретично використовуючи молекулярно-кінетичну теорію та статистичну термодинаміку. Цей коефіцієнт дещо залежить від температури, але цією залежністю можна знехтувати порівняно з e E RT .
RT .
Константа рівнянь (33-35) E має розмірність Дж/моль і носить назву енергія активації
реакції. Хоча теоретичне обґрунтування рівняння Арреніуса виконане лише для елементарних реакцій, досвід показує, що, більшість складних реакцій також підпорядковуються даному рів-
нянню. У цьому випадку, енергія активації – це мінімальна надлишкова, у порівнянні з серед-
нім рівнем, кількість енергії, яку треба надати 1 молю речовини, щоб вона вступила у хімічне перетворення.
В часи Арреніуса енергія активації розглядалась як суто емпірична константа, однак за-
раз, для елементарних реакцій, її можна оцінити з допомогою квантової хімії. Емпіричне рів-
няння (35), у наш час, також можна вивести для елементарних реакцій на основі теорії перехід-
ного стану (активованого комплексу). Згідно з цією теорією енергія активації приблизно дорів-
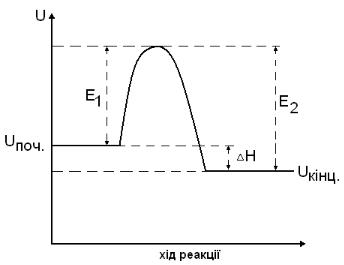
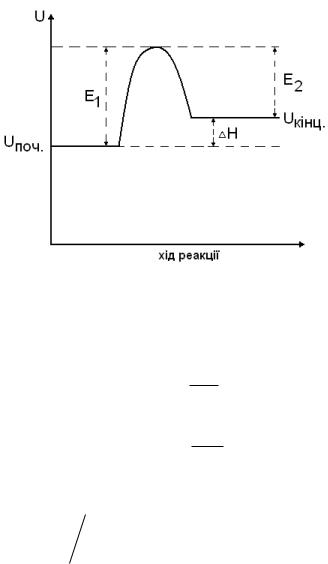
нює перевищенню середньої енергії активованого комплексу над середньою енергією вихідних речовин. Для реакції А↔В , енергії активації прямої і зворотної реакцій пов’язані співвідно-
шенням E1 E2 H . Відповідно, для екзотермічної реакції H 0 , E1 E 2 ; для ендотер-
мічної H 0 і E1 E 2 .
Рис.14. Зміна енергії під час екзотермічної реакції
© Ю.Павловський 2011
23
Рис.15. Зміна енергії під час ендотермічної реакції
Для знаходження енергії активації реакції необхідно експериментально визначити конс-
танти швидкості хоча б при двох температурах:
ln k1 ln k0 RTE1 (а), ln k2 ln k0 RTE2 (б)
Розділимо (б) на (а) і дістанемо: |
|
|
|
|
|
|
|||
|
k2 |
|
|
1 |
|
1 |
|
|
|
E R ln |
|
|
|
|
(36) |
||||
|
|
|
|||||||
|
k1 |
|
|
|
|
|
|
||
|
T2 |
|
T1 |
|
|||||
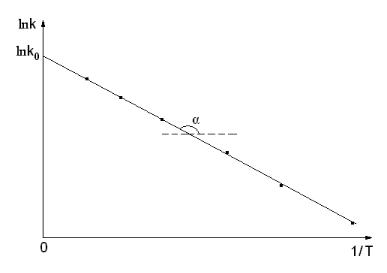
Точніше енергію активації знаходять графічно, побудувавши за експериментальними да-
ними залежність ln k f (1/ T ) . З рівняння (33) видно, що кутовий коефіцієнт отриманої пря-
мої tg E / R , а відрізок, що відсікається на осі ординат – дорівнює k0 .
© Ю.Павловський 2011
24
Рис.16. Графічне визначення енергії активації та передекспоненційного множника
Величина енергії активацій реакцій, які підпорядковуються, правилу Вант-Гоффа і про-
тікають при температурі ≈300К лежить у межах 50-100 кДж/моль.
Існують реакції для яких рівняння Арреніуса та правило Вант-Гоффа не підтверджують-
ся. Це реакції з нетермічним характером активації, гетерогенні реакції, що протікають у дифу-
зійній області, а також деякі специфічні реакції, як, наприклад, реакція окиснення оксиду азо-
ту: 2NO+O2=2NO2, для якої характерне зменшення швидкості при збільшення температури.
© Ю.Павловський 2011
25
Особливості реакцій з нетермічним характером активації
Уреакціях з термічною активацією реагуючі молекули отримують енергію активації в результаті її перерозподілу при хаотичних зіткненнях. При цьому активні молекули знаходяться
утермічній рівновазі з іншими.
Уреакціях з нетермічним характером активації необхідна енергія отримується в резуль-
таті поглинання кванту енергії при зіткненні з швидкими елементарними частинками: фотона-
ми, електронами, нейтронами, α-частинками і т.п. Такі реакції вивчає фотохімія та радіаційна хімія. Існують реакції які активуються дією електромагнітних коливань, ультразвуку і навіть механічною дією.
Для цих реакцій є декілька загальних особливостей:
1)Ці реакції протікають у дві стадії. На першій, під дією активуючого агента утворюються активні частинки: збуджені молекули, атоми, радикали, іони. Ці активні частинки взає-
модіють з іншими частинками, або з неактивними молекулами;
2)Швидкість таких реакцій не залежить від температури, бо на першу стадію температура не впливає, а друга має малу енергію активації.
3)Концентрація продуктів реакції, як правило, більша за концентрацію, що відповідає рів-
новазі при даній температурі, тому можливе протікання реакцій для яких у звичайних умовах G 0 , наприклад фотосинтез у рослинах.
Фотохімічні реакції
Фотохімічними називають реакції в яких активація частинок однієї з реагуючих речовин відбувається у результаті поглинання кванту світла.
Можна виділити дві групи таких процесів:
1)Процеси, що ініціюються світлом. Вони можуть протікати і без нього бо G 0. Світло сприяє появі активних частинок, тобто, каталізує процес. Такі реакції деколи називають фото каталітичними. Кількість речовини, що утворюється непропорційна кількості пог-
линутої світлової енергії.
2)Процеси які за даних умов (р,Т) самочинно протікати на можуть бо G 0 . Для їх про-
тікання треба затратити роботу, щоб подолання енергетичнитй бар’єр, підвівши енергію ззовні, у вигляді енергії фотонів.
Основний закон фотохімії (закон фотохімічної еквівалентності Ейнштейна): число
молекул, що вступили у первинне! фотохімічне перетворення, дорівнює числу поглинутих квантів світла.
Уфотохімічному процесі можна виділити три стадії:
©Ю.Павловський 2011

26
1)активація (первинний процес);
2)темнові процеси (вторинні);
3)дезактивація.
До первинних процесів відносять наступні:
–Збудження молекули з утворенням активної частинки здатної до наступних
перетворень:
А + hν → А*
–Дисоціація молекул на атоми або радикали:
АВ + hν → А• + В•
–Іонізація молекул з виділенням електрона:
А + hν → А+ + е
Дуже часто у первинному процесі енергію світла поглинають сторонні частинки і відда-
ють її
Вторинні процеси можуть протікати за участю дуже великої кількості молекул. Напри-
клад, коли після поглинання кванту світла, починається ланцюгова реакція. У цьому випадку на кожен поглинутий квант світла припадає багато молекул, що прореагували.
Ефективність фотохімічної реакції характеризується величиною квантового виходу:
|
число частинок, що прореаг ували |
(37) |
|
число пог линутих квантів |
|||
|
|
Якщо 1, то це пов’язано з вторинними (темновими) процесами:
1 – активні молекули встигають дезактивуватись,
1, вторинні процеси – ланцюгові.
Кількість світлової енергії, що поглинається 1 молем реагуючих речовин:
E N |
|
h N |
|
|
hc |
, |
(38) |
|
A |
A |
|
||||||
|
|
|
|
|
||||
|
|
|
|
|
|
|
де ν – частота світла, с-1;
λ – довжина хвилі світла, м;
h = 6,63*10-34 Дж*с – стала Планка;
с =3*108 м/с – швидкість світла у вакуумі.
Таблиця 4
Характеристики видимого спектру світла
Колір |
λ*10-9 м, (нм) |
ν*1012 , Гц (ТГц) |
E=hλ *10-31, Дж |
ІЧ |
>740 |
<405 |
2,69 |
Червоний |
625—740 |
480—405 |
2,69—3,18 |
Помаранчевий |
590—625 |
510—480 |
3,18—3,38 |
© Ю.Павловський 2011
27
Жовтий |
565—590 |
530—510 |
3,38—3,51 |
Зелений |
500—565 |
600—530 |
3,51—3,98 |
Блакитний |
485—500 |
620—600 |
3,98—4,11 |
Синій |
440—485 |
680—620 |
4,11—4,51 |
Фіолетовий |
380—440 |
790—680 |
4,51—5,24 |
УФ |
<380 |
>790 |
>5,24 |
З приведеної таблиці видно, що при зменшенні довжини хвилі світла енергія фотона зро-
стає. Здавалося б, що чим менша довжина хвилі, тим ефективнішою мала б бути дія світла. На-
справді ж, більшість фотохімічних реакцій є вибірковими – активуються лише світлом певної
довжини хвилі.
Дуже часто у первинному процесі енергію світла поглинають сторонні частинки і відда-
ють її учасникам реакції, а самі у реакцію не вступають (так звана фотосенсибілізація). Прик-
ладом може бути фотоліз водню УФ світлом (λ=234 нм) в присутності пари ртуті:
Hg + hν = Hg*
Hg* + Н2 = Hg + 2Н
Фотосинтез вуглеводнів рослинами у присутності хлорофілу:
6СО2 + 6Н2О + hν → С6Н12О6 + 6О2 (φ≈0,1)
Енергія активації цієї реакції 112 кДж/моль, що відповідає світлу з довжиною хвилі λ
=210 нм (жорстке УФ випромінювання), якого в спектрі сонячних променів, що досягають по-
верхні Землі немає. Хлорофіл служить тією частинкою, що поглинає світло меншої енергії (бі-
льшої довжини хвилі) і передає її учасникам реакції.
Інші приклади фотохімічних реакцій: |
|
|
1) |
утворення вітаміну в організмі |
людини D, під дією променів світла |
|
λ =310 нм на холестерол та ергостерол. |
|
2) |
утворення пероксиду водню: |
H2 + O2 + hν → H2O2 (φ=1) |
3) |
розклад сірководню у бензолі: H2S(у бензолі) + hν → H2 + S (φ=1) |
|
4) |
фотоліз йодиду водню: |
2НІ + hν → Н2 + І2 (φ=2) |
5) |
утворення озону: |
3О2 + hν → 2О3 (φ=3) |
Температурний коефіцієнт фотохімічних реакцій лежить у межах γ=1,2÷1,5, оскільки отримана в результаті поглинання світла енергія настільки велика, що підвищення температури змінити її не може.
Ланцюгові реакції
Процеси, в яких перетворення вихідних речовин у продукти реакції відбувається шляхом чергування деяких реакцій за участю вільних радикалів, називаються ланцюговими реакціями.
© Ю.Павловський 2011
28
У кожному елементарному акті ланцюгового процесу бере участь радикал, що взаємодіє з мо-
лекулою, утворюючи нові радикали.
Вільний радикал – це нестійка частинка з неспареними електронами на зовнішніх атом-
них або молекулярних орбіталях, утворена з молекули шляхом розриву одного, або декількох зв’язків.
До утворення вільних радикалів призводить: 1) дія світла:
Cl2 + hν → 2Cl•
CH3 (CO)OCH3 + hν → CH3 CO• + •OCH3
2)дія α-, β-, γ- та рентгенівського випромінювання;
3)дія ультразвуку;
4)дія електричного розряду;
5)термічний розпад:
CH3–N=N–CH3 → 2CH3• + N2 (CH3)COOH → (CH3)CO• + •OH
6) хімічні реакції з утворенням радикалів:
Fe2+ + H2O2 = Fe3+ + OH‾ + HO•
Вільні радикали живуть дуже короткий час ( наприклад для •CH3, τ=0,01 с). Їх рекомбі-
нація відбувається з виділенням дуже великої кількості енергії, яку поглинає третя частинка або стінка посудини, у якій відбувається реакція. Найбільш ймовірним процесом є зіткнення ради-
калів з молекулами реагуючих речовин і наступним їх перетворенням.
З утворенням вільних радикалів пов’язана біологічна дія різноманітних випромінювань.
Наприклад, утворені в наступному процесі радикали, реагують з ферментами, білками та пору-
шують різноманітні функції в організмі:
H2O + hν = H2O+ + e H2O+ + H2O = H3O+ + HO• H2O + e = H• + OH‾
В ланцюгових реакціях виділяють три стадії. Розглянемо їх на прикладі реакції отриман-
ня броміду водню: |
|
|
|
|
|
H2 + Br2 = 2HBr |
|
1) |
зародження ланцюга: |
Br2 + hν = 2 Br• |
|
2) |
продовження ланцюга: |
Br• + H2 |
= HBr + H• |
|
|
H• + Br2 |
= HBr + Br• |
© Ю.Павловський 2011
|
29 |
3) обривання ланцюга: |
H• + H• + M = H2 + M |
|
Br• + Br• + M = Br2 + M |
Характерною особливістю ланцюгових реакцій є залежність їх швидкості від форми по-
судини: чим менша площа поверхні посудини, тим менше сторонніх частинок і менша ймовір-
ність обриву ланцюга, та від наявності домішок.
Домішки можуть, як поглинати вільні радикали, так і сприяти їх утворенню. (див наве-
дену вище реакцію фотолізу водню у присутності пари Hg).
Прикладом може бути також реакція утворення хлориду водню
H2 + Cl2 = 2HCl:
1)в темноті ця реакція не протікає,
2)під дією світла протікає за ланцюговим механізмом з вибухом,
3)в присутності пари натрію протікає з досить великою швидкістю:
Cl2 + Na = NaCl + Cl•
Cl• + H2 = HCl + H• …
Прикладом речовин що поглинають вільні радикали та перешкоджають ланцюговим ре-
акціям окиснення в живих організмах є вітаміни C та E (так звані антиоксиданти).
© Ю.Павловський 2011
30
Кінетичні особливості гетерогенних реакцій
Гетерогенними називають реакції що протікають на межі поділу фаз. Особливістю таких реакціє є те, що в них можна виділити три характерні стадії:
1)дифузія реагентів до межі поділу фаз;
2)хімічна взаємодія (кінетичний процес);
3)дифузія продуктів реакції до межі поділу фаз.
Рис.17. Гетерогенна реакція
Загальна швидкість процесу залежить від співвідношення між дифузійним та кінетичним процесами. Якщо в системі переважає дифузія то повільнішою стадією (лімітуючою) є хімічне перетворення. Кажуть, що реакція протікає у кінетичному режимі. Якщо, навпаки, повільнішою стадією є процес масопереносу, то лімітуючою стадією є дифузія – реакція протікає в дифузій-
ному режимі.
За законом Фіка швидкість масо переносу при дифузії:
w |
dm |
|
dc |
DS |
(39) |
|
|
||||
d |
d |
|
dx |
|
|
|
|
|
|||
де dcdx - градієнт концентрації.
D kTB - коефіцієнт дифузії Енштейна (В- коефіцієнт тертя, k – стала Больцма-
на)
Замінимо dc c0 c
dx
де - така відстань, на якій концентрація змінюється від с0 на поверхні до с.
© Ю.Павловський 2011