5.2.3.1.1. Ишемическая болезнь сердца и инфаркт миокарда
Вопрос о роли эмоционального стресса в патогенезе ИБС уже давно волновал клиницистов и патофизиологов [Чазов Е.И., 1975; Lown В., De Silva R., 1978]. Это, безусловно, сложный вопрос, и чтобы определить, как стресс-реакция влияет на развитие и течение ИБС, несомненно, следовало бы вначале проанализировать патогенетическую цепь ишемического повреждения как таковую (изолированно от стресса). Однако эта проблема достаточно подробно обсуждается во всех руководствах по кардиологии и патофизиологии кровообращения, в монографиях и обзорах, поэтому мы сразу перейдем к рассмотрению роли стресса в патогенезе ишемического повреждения миокарда.
Имеющиеся данные позволяют выделить 8 основных патогенетических механизмов, посредством которых эмоциональней стресс способствует развитию ИБС и инфаркта миокарда.
Первым патогенетическим механизмом влияния стресса на патогенез ИБС являются стрессорная гиперхолестеринемия и атерогенная дислипопротеидемия.
Наблюдения клиницистов показывают, что трудная психосоциальная стрессорная ситуация, в которую попадают люди, вызывает у них подобную дислипопротеидемию, т.е. увеличение в крови уровня холестерина во фракции липопротеидов низкой плотности (ЛПНП) и рост индекса атерогенности. В экспериментах на животных было четко показано, что эмоциональный стресс, как интенсивный однократный [Твердо-хлиб В.П. и др., 1988], так и хронический [Clarkson Т.В. et al., 1987] приводят к атерогенной дислипопротеидемии, которая при хроническом стрессе сопровождается развитием коронарного атеросклероза.
Как показано в экспериментах на обезьянах, у «подчиненных» особей, т.е. занимающих в колонии подчиненное положение, что сопровождается, как известно, хроническим социальным (эмоциональным) стрессом, отношение в крови
содержания общего холестерина крови к содержанию холестерина во фракции липопротеидов высокой плотности (Хо6ш./Хлпвп) в 2 раза больше, чем у «доминирующих» особей, которые в меньшей степени подвергаются социальному стрессу. Это сопровождается у «подчиненных» обезьян увеличением частоты развития коронарных стенозов по сравнению с особями соответствующего пола, относящимися к «доминирующим». Это означает, что эмоциональный стресс приводит к увеличению в крови доли атерогенных липопротеидов и как следствие — к развитию коронарного атеросклероза [Clark-son Т.В. et al., 1987].
Основным механизмом, через который стресс приводит к атерогенной дислипопротеидемии и способствует развитию атеросклероза, можно считать стрессорное поражение печени (главного органа обмена холестерина).
В пользу этого положения свидетельствуют данные о том, что так называемая семейная гиперхолестеринемия, приводящая к коронарному атеросклерозу, связана с генетическим дефектом печени, проявляющимся в нарушении механизма захвата и катаболизма атерогенных ЛПНП [Репин B.C., Сухих Г.Т., 1998]. О поражении печени и нарушении ее функции при стрессе свидетельствуют и экспериментальные данные [Твердохлиб В.П. и др. 1988], которые показали, что при ЭБС уменьшение в крови содержания ЛПВП связано с резким падением в ней активности фермента лецитинхолестеринацетил-трансферазы, который, как известно, катализирует образование эфиров холестерина из свободного холестерина и тем самым ускоряет процесс формирования ЛПВП. Как показано, эстерифицирующая активность этого фермента через 2 ч после 6-часового ЭБС уменьшается почти в 3 раза и затем постепенно восстанавливается в течение суток. Данный фермент является короткоживущим белком, он синтезируется с высокой скоростью в печени и затем переносится в кровь, где осуществляет липопротеиновую конверсию. Поэтому постстрес-сорное падение активности фермента в крови явно указывает на нарушение его биосинтеза в печени, что может быть связано со стрессорным повреждением этого органа. На повреждение печени прямо указывает резкое повышение в крови содержания фермента фруктозо-1,6-дифосфатальдолазы, специфически отражающее повреждение печеночных клеток. Клинические данные также свидетельствуют, что стрессорная ситуация на работе приводит к нарушению функции печени у людей [Kaneko М. et al., 1996].
Повреждение печени при стрессе, очевидно, обусловлено чрезмерной активацией СРО в этом органе как результат липотропного эффекта стресс-реакции (см. раздел 5.1.5; схему 5.5). Действительно, показано, что появление в крови фрукто
зо-1,6-дифосфатальдолазы сопутствует значительной активации СРО в печени (повышению содержания малонового диа-льдегида на фоне снижения активности антиоксидантного фермента супероксиддисмутазы). Стрессорное повреждение печени приводит к нарушению в ней процессов окисления холестерина, превращения его в желчные кислоты и выведения из организма, что вызывает в свою очередь гиперхолестери-немию.
Таким образом, можно заключить, что первый патогенетический механизм влияния стресса на развитие ИБС, а именно гиперхолестеринемия и атерогенная дислипопротеидемия, обусловлен глубокими стрессорными нарушениями структуры и функции печени и нарушениями процесса удаления холестерина из организма.
В результате этого даже при отсутствии избытка холестерина в пище стресс может приводить к развитию стенозирующе-го коронарного склероза и играет важную роль в развитии ИБС.
Второй и наиболее опасный патогенетический механизм, посредством которого стресс может потенцировать тяжелые последствия ишемии и инфаркта миокарда, состоит в первичном стрессорном повреждении миокарда и активации адренергичес-ких влияний на сердце.
Первичное стрессорное повреждение сердца может иметь место до эпизода острой ишемии миокарда в результате ранее перенесенных стресс-реакций. В этом случае очаги поврежденного миокарда усугубят возникновение ишемических аритмий и нарушение сократительной функции сердца. Первичное стрессорное повреждение миокарда, т.е. повреждение некоронарного происхождения, может возникать и при ишемии в не-ишемизированных участках сердечной мышцы в результате стресс-реакции на боль и страх смерти, сопутствующих ишемии (см. раздел 5.2.3.1, схему 5.7).
Можно выделить два основных компонента отягчающего влияния стрессорного повреждения миокарда при ишемии: аритмогенный эффект стресса (усиление ишемических аритмий сердца вплоть до фибрилляции) и нарушение сократительной функции неишемизированных отделов сердца за счет стресс-индуцированного повреждения этих отделов.
Аритмогенный эффект стресса. Аритмогенное действие нейрогенного фактора при ишемии давно хорошо известно. Ишемия миокарда, вызываемая в эксперименте перевязкой левой коронарной артерии в условиях глубокого наркоза, приводила к аритмиям лишь в 6 % случаев. Стимуляция заднего гипоталамуса, осуществляющего, как известно, контроль системы кровообращения, увеличивала при этом число случаев аритмий в 10 раз [Satinsky J. et al., 1971]. Стрессорные ситуа
ции увеличивают число случаев фибрилляции желудочков при коронароокклюзии в 3 раза и в 3 раза снижают порог фибрилляции [Verrier R.L. et al., 1984]. Интересные данные были получены B.Lown и сотр. [Lown В., De Silva R., 1978]. Они использовали методику И.П.Павлова: помещали собак в станок и вырабатывали условный рефлекс на электроболевое воздействие. После этого одно помещение в станок давало стресс-реакцию, оцениваемую по росту уровня катехоламинов в крови. Такой стресс существенно снижал порог аритмий и фибрилляции сердца, т.е. повышал уязвимость сердца к аритмогенным факторам.
Аритмогенные эффекты стресса предупреждаются удалением звездчатых симпатических узлов или адреноблокатора-ми. В то же время стимуляция симпатических сердечных нервов и симпатических узлов репродуцирует аритмогенные эффекты стимуляции среднего мозга даже в условиях стабилизированных АД и частоты сердцебиений. Клинические исследования установили наличие возбуждения симпатоадреналового отдела стресс-системы во время приступов аритмии у людей, что сопровождалось увеличением экскреции катехоламинов, а также увеличением продукции цАМФ и уменьшением продукции цГМФ [Дорофеев Г.И. и др., 1985]. Таким образом, не вызывает сомнений, что возбуждение симпатических нервных центров и адренергическое влияние на сердце играют ключевую роль в патогенезе нейрогенных аритмий.
При одновременной регистрации биоэлектрической активности сердца и фронтальной коры при аритмиях, вызванных стрессом или острой ишемией, было установлено, что как действие экзогенного стрессора, так и стресс-реакция, спровоцированная ишемической болью (т.е. эндогенным стрессором), сопровождаются возбуждением определенной зоны фронтальной коры. И в обоих случаях (при стрессе и ишемии) вся цепь последующих событий, приводящих к фибрилляции сердца, оказывается кортикально обусловленной [Косицкий Г.И. и др., 1985; Skinner J.E., 1985]. Путь кора — сердце еще не весь полностью прослежен. Вместе с тем исследования на животных и клинические наблюдения показали, что фронтальная кора по меньшей мере тремя путями может регулировать состояние сердца и системы кровообращения [Косицкий Г.И. и др., 1985; Skinner J.E., 1985; Blair R.W., 1985, и др.]. Схематично эти пути в общем виде представлены на рис. 5.3.
Первый путь — кортикалъно-таламическая система, которая контролирует сенсорные каналы, т.е. выход информации от сердца и других висцеральных органов на уровень коры (I на рис. 5.3). Второй путь — фронтальная кора — лимбичес-кая система: от фронтальной коры к височной доле и ядрам амигдалы (извилина faciculus). Он обеспечивает эффекторный выход коры на ядра ствола мозга опосредованно через ядра

амигдалы,
которые в свою очередь связаны с
гипоталамусом (II на рис. 5.3). Третий,
триггерный, путь (III
на рис. 5.3), наиболее существенный для
понимания роли стресс-реакции в генезе
аритмий, связывает фронтальную кору
через субтала
мус и дорсальный гипоталамус с ядрами ствола мозга, непосредственно регулирующими систему кровообращения, т.е. кортико-стволовый путь. Данный путь регулирует висцеральные компоненты стресс-реакции, в том числе влияет на уязвимость сердца по отношению к аритмогенным факторам. Этот путь соединен обширными коллатералями с ядрами гипоталамуса (главным образом паравентрикулярным). Информация с этих коллатералей передается из гипоталамуса терминалями нейронов гипоталамуса в области ствола мозга, а именно в ядра tractus solitarius, моторные кардиоваскулярные ядра, п. ambiguus, контролирующие сердце и кровообращение. Эти же отделы гипоталамуса, т.е. получающие информацию из коры (по третьему пути), получают также терминали аксонов от амигдалы. Таким образом, гипоталамус, ключевой компонент центрального звена стресс-системы, является центром путей, реализующих аритмогенное влияние коры головного мозга при экзогенно-индуцируемом стрессе и ишемии (эндогенно вызываемом стрессе в ответ на ишемическую боль). Наиболее существенные результаты, которые выявили роль этих путей в механизме аритмий и фибрилляции сердца, были получены в исследованиях Джеймса Скиннера и его коллег из медицинского колледжа в Хьюстоне в середине 80-х годов [Skinner J.E., 1985, и др.[. Было установлено, что при окклюзии коронарной артерии и острой ишемии миокарда у свиней происходит возбуждение указанной выше зоны фронтальной коры, закономерно сопровождающееся фибрилляцией сердца. Холодовая блокада подкорковой зоны, амигдалы, а также блокада рассмотренного выше кортико-стволового пути с помощью интерцеребрального введения р-адреноблокатора пропранолола предупреждают фибрилляцию, остановку сердца и гибель животных, несмотря на ишемию миокарда.
Иными словами, было установлено, что ишемия миокарда — результат окклюзии коронарной артерии, т.е. результат механического выключения коронарного кровотока, а реакция на ишемию в форме фибрилляции и остановки сердца — результат сложных межцентральных отношений, реализующихся на уровне головного мозга.
Этот важный вывод убедительно определяет существо второго патогенетического механизма влияния стресса на развитие ишемического повреждения миокарда. Клинической иллюстрацией к этому положению служат данные о том, что у людей с повреждениями (выключением) лобных долей полностью отсутствуют вегетативные реакции на все психологически значимые стимулы (т.е. на эмоциональные стрессоры); эти люди вообще не подвержены стрессу [Tauber H.L., 1964].
Механизм аритмогенного действия стресс-реакции приближенно можно представить следующим образом. Обратимся
вновь к рис. 5.3. Стресс-реакция может возникать в ответ на внешний стрессор, а также на боль и чувство страха, сопровождающие ишемию миокарда (внутренний стрессор).
Сигнал о внешнем стрессоре воспринимается соответствующими рецепторами (обобщенно изображенными на рисунке в виде глаза) и по таламо-кортикальной системе (I путь влияния коры на сердце, см. рис. 5.3) передается в таламус и далее в воспринимающие нейроны основной коры больших полушарий, отвечающие за вход в кору. Там сигнал «переключается» и поступает во фронтальную кору. Из фронтальной коры начинается кортико-стволовый путь, как уже было показано выше (III путь на рис. 5.3), который соединяет фронтальную кору с таламусом, гипоталамусом и ядрами ствола мозга, непосредственно связанными с регуляцией сердца. Основным звеном этого пути, как указывалось выше, является гипоталамус, который «собирает» информацию от вышележащих отделов головного мозга, а также с периферии, в том числе от сердца. Из гипоталамуса информация при участии стволовых ядер — «синего пятна», п. ambiguus и др. — поступает в нейроны ядер продолговатого и спинного мозга, осуществляющих симпатическую и парасимпатическую иннервацию сердца. При этом именно преобладание симпатического выхода на сердце создает аритмогенную ситуацию. Электрическая стимуляция, или функциональная блокада этого триггерного пути, вызывает соответственно либо фибрилляцию сердца, либо ее предотвращение (в случае ишемии). На рис. 5.3 показаны варианты такой блокады, использованные в исследованиях J.E.Skinner и сотр., о которых мы говорили выше [Skinner J.Е., Reed J.С, 1981; Skinner J.E., 1985]. Холодовая блокада подкорковой зоны и амигдалы вызывает предупреждение возникновения аритмий при эмоциональном стрессе, а также предупреждает возникновение фибрилляции сердца и гибель животных при острой ишемии сердца. Интерцеребральное введение р-адреноблокаторов может воздействовать непосредственно на механизм инициации активности в триггерном пути или предупреждать переключение центральной информации в триггерном пути на вегетативный выход либо и то, и другое.
При действии внутреннего стрессора, т.е. при боли, вызванной ишемией, болевой сигнал, воспринятый рецепторами сердца, передается по афферентным волокнам, идущим от сердца в составе блуждающего нерва, а также по симпатическим волокнам, в верхние отделы ЦНС по спиноталамическо-му и спиноретикулярному трактам. Эта информация попадает в таламус и гипоталамус. Из таламуса по кортико-таламичес-кой системе, а возможно, также и непосредственно из гипоталамуса, сигнал поступает в кору больших полушарий. Далее «срабатывает» рассмотренная выше схема активации триггер-
ного фронтокортико-стволового эфферентного пути к сердцу. Кроме того, есть основания полагать, что в нейронах заднего гипоталамуса афферентная информация переключается сразу на эфферентные симпатические нейроны, аксоны которых поступают в спинальный тракт и являются преганглионарными волокнами симпатических ганглиев, иннервирующих сердце.
Разумеется, рассмотренный механизм реально намного сложнее и не все его звенья исследованы. Однако не вызывает сомнений следующее основное положение, обоснованное представленными выше исследованиями.
Стресс-реакция независимо от того, вызвана ли она внешним стрессором или внутренним, является фактором, провоцирующим аритмии и фибрилляцию сердца или отягчающим их протекание при ишемическом поражении миокарда.
Существенно, что этот механизм, первично обусловленный эмоциональной стрессорной ситуацией окружающего мира или эндогенным стрессором боли при ишемии, может «закрепиться» благодаря формированию патологической доминанты (по А.А.Ухтомскому) или патологической системы «застойно» возбужденных центров [Крыжановский Г.Н., 1997], а также благодаря возникновению при стрессе «длительного циклического движения процессов возбуждения» между кортико-лимбическими структурами [Судаков К.В., 1997а].
Следует подчеркнуть роль адренергического эффекта стресс-реакции как фактора, приводящего к возникновению или усугублению сердечных аритмий при ишемии. Во-первых, увеличение адренергического влияния на водители ритма сердца при стрессе создает преобладание адренергического влияния над холинергическим и тем самым вызывает асинхронизм в работе водителей ритма. Уже одно это может вызывать аритмии сердца вплоть до фибрилляции даже в условиях отсутствия ишемии. В условиях ишемии этот механизм повышает уязвимость сердца к ишемическим аритмиям и усугубляет тяжесть нарушений ритма вплоть до летального исхода. Во-вторых, однократно перенесенная стресс-реакция большой интенсивности или повторные эпизоды стресса за счет усиления адренергического влияния на сердце вызывают, как уже было показано выше, первичное стрессорное повреждение микроструктур сердечной мышцы и очаговые некротические повреждения. Этот комплекс изменений знаменует собой нарушение функционирования мембранного аппарата кардиоци-тов, который осуществляет генерацию и проведение возбуждения и может играть существенную роль в возникновении эктопических очагов, из которых исходят «преждевременные» импульсы возбуждения, и очагов функционального блока проведения, что при инфаркте и острой ишемии может приводить
18—1385
273
к возникновению хорошо известного феномена reentry — признанную основу желудочковой тахикардии и фибрилляции сердца.
Нарушение сократительной функции неишемизированных отделов миокарда. При описании первичного стрессорного повреждения выше было показано, что это повреждение характеризуется постстрессорной ригидностью сердечной мышцы и снижением ее сократительной функции. Оказалось, что при экспериментальном инфаркте миокарда левого желудочка у крыс, вызываемого, по Г.Селье, перевязкой левой нисходящей коронарной артерии, в неишемизированных отделах сердца (предсердиях) также развиваются ригидность, нарушение сократительной функции и снижение резистентности к высоким концентрациям кальция и гипоксии. Иными словами, при ишемическом повреждении сердца в неишемизированных отделах возникают те же нарушения функции и структуры сердечной мышцы, что и в интактном сердце после интенсивного стрессорного воздействия. Предварительное введение р-блокатора индерала, предупреждающее избыточное адренергическое влияние на сердце при стрессе, в высокой степени защищает сократительную функцию неишемизированных предсердий при инфаркте левого желудочка. Этот защитный эффект, как и в случае ЭБС, выражался в значительном уменьшении ригидности и снижения сократительной функции предсердия, а также в повышении его резистентности к гипоксии и перегрузке кальцием (см. раздел 5.2.3.1).
Таким образом, при ишемическом поражении сердца стресс-реакция, вызываемая внешним стрессором (окружающей средой) или внутренним стрессором (болью в сердце и страхом смерти), является патологическим фактором, вызывающим или потенцирующим нарушения ритма сердца и депрессию сократительной функции неишемизированных отделов миокарда.
Третий патогенетический механизм, через который реализуется роль стресса в патогенезе ИБС и инфаркта миокарда, состоит в том, что сильный адренергический компонент стресс-реакции, сопутствующей острой ишемии, может приводить к спазму гладкой мускулатуры анатомически интактных коронарных артерий, и этот достаточно стойкий спазм становится причиной вторичного ишемического поражения миокарда.
При рассмотрении адаптивных эффектов стресс-реакции мы отмечали, что вызываемая катехоламинами через р-адре-норецепторы сосудов «рабочая гиперемия» миокарда (дилата-ция коронарных сосудов) при увеличении интенсивности и длительности стресс-реакции может сменяться коронарокон-стрикцией. Это происходит в связи с десенситизацией р-адре-норецепторов большими дозами катехоламинов и реализацией эффекта катехоламинов через констрикторные а|-адреноре-
цепторы. В этом смысле очень показательны исследования [Simons М., Downing S.E., 1985], в которых длительно вводили животным внутривенно НА и регистрировали коронарный кровоток. Авторы показали, что вначале в ответ на НА кровоток увеличивался и возвращался к исходному уровню, а затем сопротивление коронарного русла возрастало вдвое, кровоток снижался, и 48 ч спустя развивалось ишемическое повреждение сердца. При этом на фоне блокатора а-адренорецепторов фентоламина коронароспазм и ишемическое поражение не возникали.
Важное значение в регуляции коронарного кровотока, как показывают исследования последних 5 лет, имеет NO — мощный вазодилататор. При умеренной стресс-реакции продукция NO возрастает [Малышев И.Ю., Манухина Е.Б., 1998], и это играет ключевую роль в реализации «рабочей гиперемии». При длительном и интенсивном стрессорном воздействии генерация NO может падать, и этот фактор, очевидно, — одна из решающих причин возникновения стрессорного коронароспаз-ма. Поэтому при стенокардии помогают нитроглицерин и другие нитропрепараты, являющиеся донорами NO.
Установлено, что длительный ЭБС вызывает у крыс существенное уменьшение отношения содержания в крови корона-родилататора простациклина (ПП2), коронароконстриктора и стимулятора тромбообразования тромбоксана А2 (ТХА2), т.е. ПП2/ТХА2, а также отношения коронародилататора ПГЕ, и коронароконстриктора nrF2a, т.е. ПГЕ,/П1Т2а. Эти изменения могут играть роль в возникновении или потенциации окклюзии коронарных сосудов и усугублении ишемии [Пшенни-кова М.Г. и др., 1992].
Таким образом, стресс создает благоприятные условия для возникновения коронароспазма. Врожденная сниженная активность стресс-лимитирующих систем предрасполагает к стрессорным повреждениям, в том числе к нарушению коронарного кровотока при стрессе. Напротив, повышение мощности этих систем является важным фактором профилактики стрессорных повреждений и стресс-индуцированного усугубления ИБС.
Четвертый патогенетический механизм влияния стресса на патогенез ИБС и инфаркта миокарда состоит в том, что стресс-индуцированный «выброс* катехоламинов в кровь потенцирует свертывание крови и тромбоз коронарных сосудов. Следствием возникающей при этом агрегации тромбоцитов является выделение из них мощных вазоактивных веществ, особенно ТХА2, серотонина и гистамина, которые заведомо усиливают спазм и в сочетании с тромбозом делают его более опасным. Это имеет место чаще всего в тех зонах коронарного русла, где изначально имеются атеросклеротические изменения. Действительно, клинические наблюдения свидетельствуют о том,
18»
275
что для больных коронарной болезнью интенсивный эмоциональный («ментальный») стресс очень опасен: он может вызывать ишемию и инфаркт миокарда. Так, холтеровское монито-рирование ЭКГ у 112 мужчин и 14 женщин (средний возраст 56 лет), страдающих коронарной болезнью, показало, что ментальный стресс вызывал у них резкие ишемические изменения [Morris J.J. et al., 1996]. При этом важную роль в роковом стрессорном усугублении коронарной болезни играет стресс-индуцированный тромбоз. У военных летчиков один ответственный полет на истребителе вызывает повышение активности тромбина в 2 раза [Biondi G. et al., 1996].
Как известно, сильным противодействием тромбозу обладают простациклин и ПГЕ. Они ингибируют агрегацию тромбоцитов, и, таким образом, стресс-лимитирующая система протекторных ПГ является не только противоконстрикторной, но и антитромбозной. Указанное выше стресс-индуцирован-ное уменьшение отношений ППг/ТХАг и ПГЕ|/ПГР2а не только способствует коронароспазму, но и, несомненно, потенцирует тромбообразование или даже обусловливает его возникновение при ишемии.
Острая ишемия и инфаркт миокарда могут быть результатом сочетанного эффекта таких быстродействующих патогенетических механизмов, как спазм и тромбоз, и органических изменений, вызванных медленно развивающимся атеросклерозом.
На основании изложенного это означает, что при одинаковом атеросклеротическом коронаростенозе острая ишемия может возникать или, напротив, отсутствовать в зависимости от состояния нейрогуморальной регуляции сосудов, т.е. от активности стресс-системы и стресс-лимитирующих систем. Стресс-реакция достаточной интенсивности на фоне дефицита системы протекторных ПГ и снижения генерации NO будет сильным потенциатором возникновения острой ишемии. Напротив, активация системы протекторных ПГ и увеличение мощности системы генерации NO могут способствовать уменьшению угрозы ишемического повреждения сердца при коронаростенозе.
Таким образом, вопрос о том, возникнет ли стойкий спазм, тромбоз и в конечном счете инфаркт миокарда, в высокой степени зависит от состояния таких локальных стресс-лимитирующих систем, как системы протекторных ПГ и NO.
Пятый патогенетический механизм, через который реализуется роль стресса в патогенезе ишемического повреждения сердца, формируется на основе такого компонента стресс-реакции, как гипервентиляция. Гипервентиляция закономерно влечет за собой увеличение напряжения кислорода в крови и гипокапнический алкалоз. Эти сдвиги в свою очередь умень
шают коронарный кровоток и повышают тонус коронарных сосудов в связи со снижением в крови содержания Н+, которое приводит к увеличению связывания Са2+ с тропонином в миофибриллярном аппарате миоцитов сосудистой стенки с последующим их констрикторным сокращением. Гипервентиляция и сопутствующий ей гипокапнический алкалоз вызывают у больных с ИБС уменьшение коронарного кровотока и появление приступов стенокардии [Groves В. et al., 1977]. Характерно, что эти приступы купировались введением Са2+-блокатора дилтиазема, который не влиял на газовый состав крови в коронарном русле, т.е. парциальное давление углекислоты оставалось сниженным, а кислорода — увеличенным. Эти данные не только указывают на роль стрессорной гипервентиляции в обострении ИБС, но и позволяют вновь подчеркнуть ключевую роль кальция в механизме повреждения.
Шестой патогенетический механизм состоит в том, что длительная стресс-реакция снижает резистентность миокарда к гипоксии и реоксигенации. Это положение было доказано в исследованиях, где одновременно оценивались сократительная функция сердца, периферическое сопротивление в сосудах, АД, а также ударный и минутный объемы сердца и его работа [Белкина Л.М. и др., 1983, и др.]. Оценку состояния сердца и кровообращения осуществляли в условиях нормоксии, 30-минутной ишемии и последующей реоксигенации сердца у животных, предварительно перенесших ЭБС (за 2 ч до тестирования). Оказалось, что у перенесших ЭБС животных в условиях нормоксии наблюдалось лишь незначительное статистически недостоверное ухудшение перечисленных параметров функции сердца. Иное отмечено в условиях ишемии и особенно реоксигенации сердца: предварительно перенесенный стресс значительно увеличивал вызываемое ишемией падение систолического напряжения левого желудочка. В еще большей степени перенесенный стресс потенцировал реоксигенационные (постишемические) нарушения сократительной функции сердца.
Таким образом, стресс является фактором, отягчающим повреждение сердца при ишемии и реоксигенации.
Седьмой патогенетический механизм, благодаря которому стресс усугубляет ишемическое поражение сердца, состоит в адренергической мобилизации сократительной функции сердечной мышцы. В сочетании с регуляторно обусловленным повышением сопротивления сосудистого русла это создает значительную нагрузку на сердце и может существенно потенцировать ишемическое повреждение не только при спазме или тромбозе, но и при «простом» атеросклеротическом стенози-ровании коронарных сосудов.
На первый взгляд, это явление, казалось бы, противоречит тому, что р-адренергический эффект стресса первично приводит к коронарной вазодилатации. G.H.Mudge и соавт. (1976)
объясняли это явление тем, что в условиях предшествующего стеноза крупной коронарной артерии дополнительная нагрузка не может быть компенсирована за счет регуляторного расслабления лежащей ниже стеноза области коронарного русла, так как достаточный объем кровотока в этой области все равно не может быть достигнут из-за уменьшенного стенозом притока. Это объяснение, хотя и неисчерпывающее, согласуется с очень «красивыми» данными, полученными R.L.Verrier и соавт. (1984) при действии эмоционального стресса на собак с вживленной манжетой на коронарной артерии. Они показали, что стресс, вызываемый отнятием пищи, приводил у животных без стеноза коронарной артерии к р-адренергическим эффектам, т.е. к увеличению коронарного кровотока и падению сопротивления в коронарном русле. У животных, которым создавали стеноз коронарной артерии пережатием манжеты, такой стресс, напротив, приводил к увеличению сопротивления в коронарном русле, что свидетельствовало о стресс-ин-дуцированной коронароконстрикции.
Таким образом, несмотря на всю сложность механизма рассматриваемых явлений, факт остается фактом — стресс может усугублять ишемическое поражение сердца, вызывая ад-ренергическую мобилизацию его функции.
Восьмой патогенетический механизм, посредством которого стресс-реакция может вызывать нарушение кровообращения и приводить к ишемии не только сердца, но и мозга, состоит в выраженном нарушении сократительной функции и тонуса сосудов. В основе этого положения лежат данные, полученные в последние годы в экспериментах на изолированных венах и аорте. Во-первых, было показано, что стресс приводит к уменьшению сократительной функции мускулатуры воротной вены и снижению ее адренореактивности, причем подобное нарушение сократительной функции воротной вены возникает и при инфаркте миокарда. В данном случае решающим фактором, по-видимому, является стресс-реакция, возникающая в ответ на боль [Манухина Е.Б., 1988]. Таким образом, стресс-индуцированное снижение сократительной функции портальной вены может играть роль в избыточном кровенаполнении портального русла, возникновении артериальной гиповолемии и коллаптоидных состояний при тяжелом стрессе и инфаркте миокарда. Во-вторых, было установлено, что при эмоциональном стрессе и экспериментальном инфаркте миокарда [Manukhina Е.В. et al., 1996] происходят падение тонуса артериальных сосудов, снижение АД, сопровождающееся уменьшением чувствительности сосудов к вазоконстрикторным факторам. Как известно, выраженная гипотензия и гипореак-тивность сосудов в ответ на констрикторные (адренергичес-кие) стимулы составляют важное звено в патогенезе кардио-генного шока в острый период инфаркта миокарда.
При изучении механизма падения тонуса сосудов и АД при стрессе и инфаркте миокарда было обнаружено, что ключевая роль в этом явлении принадлежит гиперактивации эндотелия сосудов, которая приводит к увеличению расслабления гладкой мышцы сосудов и подавлению адренергичес-ких вазоконстрикторных реакций. Выяснено, что это угнетающее действие эндотелия на сократительную функцию сосудов и их реакцию на вазоконстрикторы связано с продукцией эндотелием N0. Действительно, при инфаркте миокарда было доказано увеличение синтеза N0 в организме [Va-nin A.F. et al., 1994].
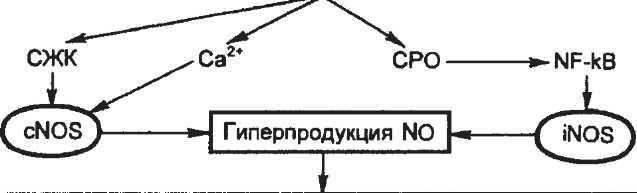
Механизм стимуляции гиперпродукции N0 при острой ишемии и инфаркте миокарда остается во многих аспектах неясным. С современных позиций можно полагать, что важную роль в этом механизме играют активация липидной триады, СРО и соответственно увеличение содержания в миоцитах сосудистой стенки Са2+, свободных радикалов и жирных кислот. Эти события вызываются самой ишемией миокарда и в еще большей степени стресс-реакцией, возникающей в ответ на ишемическую боль и страх смерти и на внешние стрессоры, т.е. сопутствующую стрессорную ситуацию окружающей среды. Увеличенная концентрация свободных жирных кислот, увеличенная концентрация внутриклеточного Са2+ и свободные радикалы вызывают активацию NO-синтазы, ключевого фермента продукции NO и рост этой продукции. Данный процесс можно представить в виде гипотетической схемы, приведенной на схеме 5.8. Схема показывает, что стресс-реакция, вызывая мощный липотропный эффект, рассмотренный выше (см. раздел 5.1.5), приводит к накоплению указанных факторов, которые активируют NO-синтазы: конституитивную N0-синтазу (cNOS) и индуцибельную, т.е. рецепторзависимую, NO-синтазу (iNOS). Как следствие возникает гиперпродукция N0, и в результате всей цепочки процессов могут развиваться гипотензия, недостаточность кровообращения и кардиоген-ный шок.
Таким образом, гиперпродукция NO и развивающееся в результате уменьшение периферического сопротивления сосудов, а также уменьшение реактивности сосудов к контрак-тильным факторам, приводящие к острой гипотензии, являются важным патогенетическим механизмом, посредством которого стресс может усугублять течение острой ишемии.
В целом рассмотренные патогенетические механизмы, через которые реализуется роль стресса в патогенезе повреждений сердца при ишемии и инфаркте миокарда, обобщены на схеме 5.9.
Стресс-реакция является чрезвычайно важным фактором возникновения, течения и исхода ИБС. Поэтому для рацио-
Схема 5.8. Патогенетический механизм, посредством которого стресс-реакция при острой ишемии миокарда может вызвать недостаточность кровообращения и кардиогенный шок [модификация схемы: Manukhina Е.В. et al., 1996]
Острая ишемия миокарда] \ Внешние стрессоры \
Боль, страх
Стресс-реакция^-