

4.3.6. Апоптоз
Апоптоз (или программируемая клеточная гибель) — широко распространенный биологический феномен клеточного «самоубийства», которое индуцируется либо разнообразными внешними стимулами, либо неразрешимыми «внутренними» конфликтами (например, невозможностью репарации повреждений ДНК). Роль апоптоза велика не только в формообразо
вательных процессах во время эмбриогенеза (формирование органов, замена одних тканей другими, резорбция временных органов и т.д.), но и в поддержании тканевого гомеостаза во взрослом организме. В регуляции тканевого гомеостаза клеточная гибель выполняет функцию, комплементарную митозу. У опухолевых клеток программа клеточной гибели во многих случаях блокирована, что вносит существенный вклад в увеличение массы опухоли.
Феноменология. Существует два типа клеточной гибели: некроз и апоптоз (клеточное «убийство» и «самоубийство» соответственно), различающиеся как индуцирующими эти процессы стимулами, так и механизмом их осуществления.
Некроз — метаболическая катастрофа, вызванная тяжелыми повреждениями клеточных структур. Для него характерно раннее увеличение объема клетки и митохондрий с последующим их автолизом, тогда как внутриядерные нарушения обнаруживаются позднее (таким образом, вектор видимых изменений направлен от периферии клетки к ее центру — ядру).
Апоптоз как явление физиологическое распространен гораздо шире. С его участием проходят морфообразовательные процессы в эмбриогенезе, позитивная и негативная селекция Т- и В-лимфоцитов, индуцированная глюкокортикоидами гибель лимфоцитов, а также гибель клеток при их естественном старении. Усиленный апоптоз ведет к инволюции органа, что встречается и в физиологических условиях (например, молочная железа в постлактационном периоде), и при патологии (например, при дегенеративных заболеваниях центральной нервной системы).
Опухолевые клетки имеют селективное преимущество перед нормальными, поскольку механизм апоптоза у них зачастую дефектен (в частности, о выживании опухолевых клеток в условиях гипоксии см. далее). Кроме того, у многих из них развивается резистентность к различным химиопрепара-там и к облучению, которые индуцируют гибель также по механизму апоптоза. Морфологически апоптоз проявляется вначале в конденсации и маргинации хроматина, уменьшении объема клетки и фрагментации клеточного ядра, в характерной складчатости плазматических мембран (вектор изменений — от центра клетки к периферии). Одним из ключевых моментов апоптоза, знаменующим необратимость процесса, является распад ДНК. Процесс заканчивается фрагментацией клетки с образованием так называемых апоптотических телец, которые быстро фагоцитируются макрофагами или соседними клетками без сопутствующей воспалительной реакции.
Механизмы апоптоза. Прорыв в выяснении механизмов апоптоза у млекопитающих был достигнут в результате изучения весьма, казалось бы, экзотического объекта — нематоды Caenorhabditis elegans, на определенной стадии развития кото
рой в строгой очередности погибает 131 соматическая клетка из 1090 имеющихся [Horvitz, 1986]. Эта оказавшаяся исключительно ценной модель позволила идентифицировать более десятка участвующих в апоптозе генов. В частности, ced-З и ced-4 (cell death abnormal) необходимы для клеточной гибели и при их дефектах клетки не погибают; напротив, ced-9 кодирует белок, предотвращающий апоптоз (его мутации сопряжены с массовой клеточной гибелью). Продукт гена ced-З, как оказалось, является протеазой.
Принципиальное значение имел тот факт, что механизмы апоптоза чрезвычайно консервативны и сохраняют свои фундаментальные черты у весьма далеких в эволюционном отношении организмов. Это обстоятельство позволило идентифицировать у млекопитающих (и, в частности, у человека) гены, гомологичные генам апоптоза у нематоды.
Гомологи ced-9 — члены семейства bcl-2. У млекопитающих гомологами ced-9 являются члены семейства bcl-2 [ген bcl-2 впервые обнаружен в участке хромосомной транслокации t(14; 18) в В-клеточной фолликулярной лимфоме]. Его роль антиапоптическая, он предотвращает гибель клеток. Белок, кодируемый bcl-2 (мол. масса 26 кДа), локализован преимущественно в мембране митохондрий и не содержит последовательностей с уже известными каталитическими или иными функциями. Фосфорилирование серина в Bcl-2 инактивирует его и может, таким образом, играть регуляторную роль.
Механизм апоптоза у млекопитающих неизмеримо сложнее, чем у нематод (анализ генов последних был лишь начальным, хотя и очень важным толчком). Так, у bcl-2 млекопитающих имеется множество гомологов, образующих семейство и по-разному участвующих в программируемой клеточной гибели. Все белки этого семейства объединены общностью структуры — присутствием функционально важных областей ВН1, ВН2 и ВИЗ (Bcl-2 homology regions 1, 2 и 3), но могут быть подразделены на две группы соответственно их антиапоптическому (Bcl-2, BHRF1, MCL-1, Bcl-xL) или про-апоптическому (Вах, Bcl-xs, Bak) действию. Белок-супрессор р53 активирует Ьах и ингибирует bcl-2-гены, что, возможно, является механизмом апоптоза, индуцируемого у клеток с поврежденной ДНК на границе G1/S (см. выше). Вместе с тем необходимо заметить, что индукция апоптоза может происходить как опосредованно через р53, так и независимо от него.
Члены семейства способны к гомо- и гетеродимеризации. Так, Вах образует гомодимеры и гетеродимеры с Bcl-2, Bcl-xL и другими белками. Комплексирование проапоптических и антиапоптических белков ведет, по-видимому, к их взаимному «погашению», в результате чего конечный результат (индукция апоптоза или, напротив, его отмена) определяется «балан-
Схема 4.3. Взаимоотношения семейства Bcl-2 и семейства ICE-подобных протеаз при индукции апоптоза
Сигнал выживания
Сигнал гибели

т

'Активация ICE-подобных протеаз
1
![]()
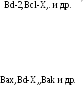
Расщепление
специфических
субстратов
(ламины, PARP,
RNP и др.)
Апоптоз
Ламины — белки, составляющие основу ядерной мембраны; PARP — poly (ADP-ribose) polymerase, фермент, участвующий в репарации ДНК; RNP — рибонуклеопротеидные комплексы. А — активация; И — ингибирова-ние.
сом сил». Отношение Bcl-2 к Вах определяет чувствительность данной клетки к апоптическим стимулам (в связи с этим говорят о реостате Bcl-2/Вах). Поскольку гибель опухолевых клеток под воздействием химиопрепаратов и облучения происходит по механизму апоптоза, то резистентность опухолей к этим воздействиям может быть обусловлена повышенной экспрессией bcl-2.
Гомологи ced-З — члены семейства протеаз. У млекопитающих гомологом гена нематоды ced-З является ген специфической цистеиновой протеазы ICE, расщепляющей предшественник интерлейкина-ip. Его искусственная (в условиях генно-инженерных экспериментов) гиперэкспрессия в культивируемых клетках вызывает апоптоз. iCE функционирует в виде тетрамера двух субъединиц размером 10 и 20 кДа и имеет несколько гомологов с разной субстратной специфичностью. Хотя участие ICE в апоптозе несомненно, мишени ее протео-литической активности пока не определены.
Функциональные взаимоотношения между семейством Bcl-2 и семейством ICE-подобных протеаз в настоящее время не установлены окончательно, хотя в апоптических сигнальных путях первые явно стоят «выше по течению» (схема 4.3).
Fas/APO-1 (CD95) — рецепторы специфических индукторов апоптоза. Большое функциональное значение имеет механизм апоптоза, индуцируемого через специфические рецепторы — CD95 (трансмембранный белок-рецептор размером 45 кДа, который при связывании со специфическим лигандом или антителами передает сигнал к апоптозу) и TNF-R (tumor necrosis factor receptor — рецептор фактора некроза опухолей).
Эти рецепторы, объединяемые сходством внеклеточных доменов, входят в состав большого семейства.
Лигандами (молекулами, специфически взаимодействующими с рецепторами TNF-R и CD95), являются соответственно TNF и CD95-L, которые представляют собой трансмембранные белки, но могут функционировать и в растворимой, «свободной» форме. Особенно интересен с онкологической точки зрения TNF — цитокин, производимый многими клетками (макрофагами, моноцитами, лимфоидными клетками, фибробластами) в ответ на воспаление, инфекцию и другие стрессорные воздействия. Он индуцирует широкий спектр иногда противоположных по направленности реакций, включая лихорадку, шок, некроз опухоли, анорексию; индуцирует иммунорегуляторные сдвиги, клеточное размножение, дифференцировку и апоптоз. Эффекты TNF опосредованы через два рецептора (трансмембранные белки размером 55 и 75 кДа, TNF-RI и TNF-RI I соответственно), по крайней мере один из которых присутствует во всех клетках.
Взаимодействие трех молекул TNF с TNF-RI «сшивает» рецепторы с образованием тримера, что запускает каскад последующих разветвленных реакций, ответственных за плейо-тропные эффекты TNF. Их непосредственный итог зависит от того, какие именно сигнальные белки окажутся рекрутированными в состав первичного комплекса. Тримеризация рецептора дает начало самосборке под плазматической мембраной сложной конструкции, существующей благодаря белок-белковому взаимодействию «стыковочных узлов», получивших название «доменов смерти» (death domains), — специфической последовательности, присутствующей во внутриклеточной части рецептора и абсолютно необходимой для передачи цито-токсического сигнала (рис. 4.15).
В комплекс рекрутируются белки TRADD (TNFRI — associated death domain protein), FADD/MORT1 (Fas — associated protein with death domain) и RIP (receptor — interacting protein), С-концевые участки которых содержат «домены смерти» (RIP, кроме того, является серин/треониновой киназой). В комплексе также участвуют TRAF1 и TRAF2 (TNFR — associated proteins 1 и 2). В свою очередь N-концевой домен FADD взаимодействует с ICE-подобной протеазой MACH (MORT1 — associating CED homolog), известной и как FLICE (FADD-like ICE), непосредственно запускающей апоптический протеаз-ный каскад. В то время как FADD включает механизм апоптоза, другие участники комплекса индуцируют иные эффекты: TRAF2 и RIP активируют NF-кВ (транскрипционный фактор плейотропного действия, оказывающий антиапоптическое действие) и JNK (см. выше).
Активация комплекса NF-кВ происходит своеобразно: фосфорилирование ингибирующей субъединицы 1кВ приво-

Анти-апоптоз
JNK
Рис. 4.15. Модель рекрутирования сигнальных молекул при активации TNF-RI, индуцирующей апоптоз и другие клеточные реакции. Вызываемая лигандом тримеризация TNF-RI ведет к рекрутированию TRADD (путем связывания с цитоплазматическим доменом рецептора через «домены смерти» — полностью заштрихованные участки). TRADD служит платформой для связывания двух дополнительных белков, обладающих «доменами смерти», — RIP и FADD, а также TRAF2, который не обладает этим доменом; в последнем случае взаимодействуют N-концевая последовательность TRADD и С-кон-цевая последовательность TRAF2 (частично заштрихованный участок). В то время как FADD индуцирует апоптический протеазный каскад, TRAF2 и RIP активируют транскрипционный фактор широкого спектра действия NF-kZ? (оказывает антиапоптическое действие) и киназный каскад. Пояснения в тексте.
дит к ее высвобождению из цитоплазматического комплекса, включающего также ДНК-связывающие субъединицы р50 и р65 (последняя известна также как Rel-A). Гетеродимер p50/Rel-A транслоцируется затем в ядро клетки, где связывается со специфической последовательностью ДНК.
Таким образом, тримеризация TNF-RI ведет к активации по крайней мере трех сигнальных путей, что объясняет разнообразие биологических эффектов TNF.